SparseAssembler2: Sparse k-mer Graph for Memory Efficient Genome Assembly
The formal version of our work has been published in BMC Bioinformatics and can be found here: http://www.biomedcentral.com/1471-2105/13/S6/S1 Motivation: To tackle the problem of huge memory usage associated with de Bruijn graph-based algorithms, upon which some of the most widely used de novo genome assemblers have been built, we released SparseAssembler1. SparseAssembler1 can save as much as 90% memory consumption in comparison with the state-of-art assemblers, but it requires rounds of denoising to accurately assemble genomes. In this paper, we introduce a new general model for genome assembly that uses only sparse k-mers. The new model replaces the idea of the de Bruijn graph from the beginning, and achieves similar memory efficiency and much better robustness compared with our previous SparseAssembler1. Results: We demonstrate that the decomposition of reads of all overlapping k-mers, which is used in existing de Bruijn graph genome assemblers, is overly cautious. We introduce a sparse k-mer graph structure for saving sparse k-mers, which greatly reduces memory space requirements necessary for de novo genome assembly. In contrast with the de Bruijn graph approach, we devise a simple but powerful strategy, i.e., finding links between the k-mers in the genome and traversing following the links, which can be done by saving only a few k-mers. To implement the strategy, we need to only select some k-mers that may not even be overlapping ones, and build the links between these k-mers indicated by the reads. We can traverse through this sparse k-mer graph to build the contigs, and ultimately complete the genome assembly. Since the new sparse k-mers graph shares almost all advantages of de Bruijn graph, we are able to adapt a Dijkstra-like breadth-first search algorithm to circumvent sequencing errors and resolve polymorphisms.
💡 Research Summary
**
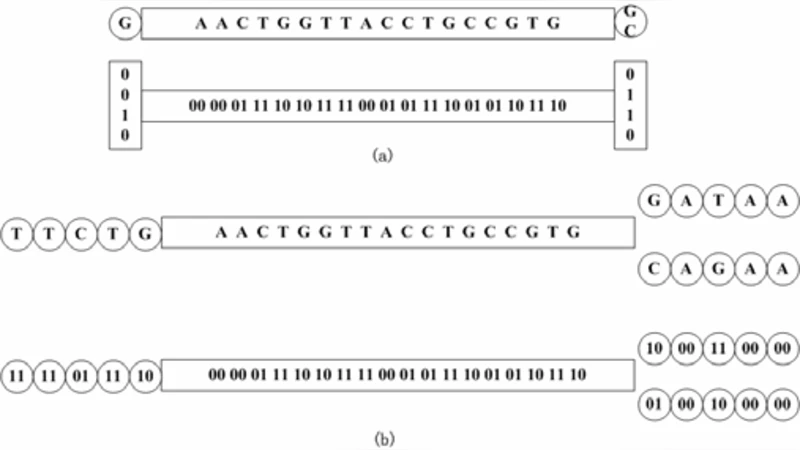
SparseAssembler2 introduces a fundamentally different approach to de novo genome assembly that addresses the severe memory consumption of traditional de Bruijn‑graph‑based assemblers. Instead of storing every overlapping k‑mer extracted from sequencing reads, the method selects a sparse subset of k‑mers at a user‑defined interval g (typically 16–25). These selected k‑mers become the graph nodes, and the links between them are inferred from the reads, effectively compressing the intervening (g – 1) bases into a compact edge representation.
The construction proceeds in two passes. In the first pass, each read is scanned; if a previously selected node is encountered within the next g k‑mers, the scan jumps to that node, otherwise the current k‑mer is added as a new node. After this pass the nodes are roughly g‑spaced, and low‑coverage nodes—likely arising from sequencing errors or spurious branches—are filtered out. In the second pass, the algorithm revisits all reads to establish directed links between the surviving nodes, recording the compressed nucleotide sequences that bridge them. The resulting sparse k‑mer graph contains only about 1/g of the original k‑mers, and each node stores 24 bits for the k‑mer itself plus 2 × g bits for the edge information, plus a modest pointer overhead. Consequently, memory usage drops by roughly an order of magnitude compared with a full de Bruijn graph, while still preserving the essential connectivity needed for assembly.
Error handling is integrated directly into the graph building and traversal stages. After the second pass, weak links (those supported by few reads) are removed. Then a Dijkstra‑like breadth‑first search is applied: when a visited node is encountered again, the algorithm backtracks to the last branching point and continues along the higher‑coverage branch, discarding tips, bubbles, and tiny loops. This strategy eliminates most error‑induced structures without requiring separate denoising steps, a major improvement over SparseAssembler1, which needed multiple rounds of error correction.
Performance was evaluated on simulated and real datasets. In noise‑free simulations of Drosophila melanogaster, Oryza sativa, and Escherichia coli genomes, SparseAssembler2 consistently outperformed SOAPdenovo, Velvet, and ABySS in terms of memory consumption (≈ 10 % of the memory used by competitors) while achieving comparable or superior N50 and longest contig lengths. When realistic substitution errors (0.5 %–2 %) were introduced into the E. coli reads, the assembler still produced reasonable assemblies, demonstrating robustness to sequencing errors.
A real‑world test on a 416 Mbp Illumina dataset from the bee Lasioglossum albipes (60× coverage) highlighted the practical benefits: SparseAssembler2 required only 3.5 GB of RAM versus ~30 GB for SOAPdenovo, completed the job faster, and yielded a slightly higher N50 (1790 bp vs. 1689 bp) and total assembled length (262 Mbp vs. 258 Mbp).
In summary, SparseAssembler2 replaces the exhaustive de Bruijn graph with a sparse k‑mer graph that stores far fewer nodes and compact edge information, integrates error correction into graph traversal, and achieves dramatic memory savings without sacrificing assembly quality. The approach opens the possibility of assembling moderate‑to‑large genomes on standard desktop computers and suggests future extensions such as adaptive g‑spacing, multi‑k strategies, or integration with succinct data structures for even greater efficiency.
Comments & Academic Discussion
Loading comments...
Leave a Comment