CplexA: a Mathematica package to study macromolecular-assembly control of gene expression
Summary: Macromolecular assembly vertebrates essential cellular processes, such as gene regulation and signal transduction. A major challenge for conventional computational methods to study these processes is tackling the exponential increase of the number of configurational states with the number of components. CplexA is a Mathematica package that uses functional programming to efficiently compute probabilities and average properties over such exponentially large number of states from the energetics of the interactions. The package is particularly suited to study gene expression at complex promoters controlled by multiple, local and distal, DNA binding sites for transcription factors. Availability: CplexA is freely available together with documentation at http://sourceforge.net/projects/cplexa/.
💡 Research Summary
The paper introduces CplexA, a Mathematica package designed to tackle the combinatorial explosion that arises when modeling macromolecular assemblies involved in gene regulation. In many biological systems, a promoter may contain dozens of transcription‑factor (TF) binding sites, each of which can be either occupied or free, and TFs can interact cooperatively or antagonistically over long distances. The total number of possible configurations therefore grows as 2^N, where N is the number of sites, quickly rendering exhaustive enumeration infeasible for conventional computational approaches.
CplexA circumvents this problem by exploiting functional programming constructs native to Mathematica. The user supplies thermodynamic parameters: binding free energies for each TF‑site pair, interaction energies between TFs (including cooperative loops, distal contacts, and chromatin‑mediated effects), and a mapping from each configuration to an observable (e.g., transcriptional output). The package then constructs a symbolic representation of the state space as a list of binary occupation vectors. Rather than iterating over all 2^N vectors, CplexA uses recursive convolution‑type operations to compute the total free energy of each configuration, applies the Boltzmann factor exp(−ΔG/RT) to obtain unnormalized weights, and finally normalizes them to yield exact probabilities for every state. This algorithm reduces the effective computational complexity from exponential to linear in the number of interacting clusters, making it possible to analyze systems with dozens of sites on a standard desktop.
The core functionality is organized into four high‑level functions: (1) DefineParameters – creates a data structure containing all binding and interaction energies; (2) ComputeEnergy – evaluates the total free energy for any given occupation vector; (3) PartitionFunction – performs the Boltzmann sum over the implicit state space; (4) Expectation – calculates ensemble averages of user‑specified observables such as promoter occupancy, cooperative pairwise contact probabilities, or the mean transcription rate. Because these functions are built from Mathematica’s symbolic and list‑processing primitives, they can be combined with custom user code, allowing the modeling of sophisticated logical gates (AND, OR, NAND) within a promoter, the inclusion of epigenetic modifiers, or the coupling of transcription to signaling cascades.
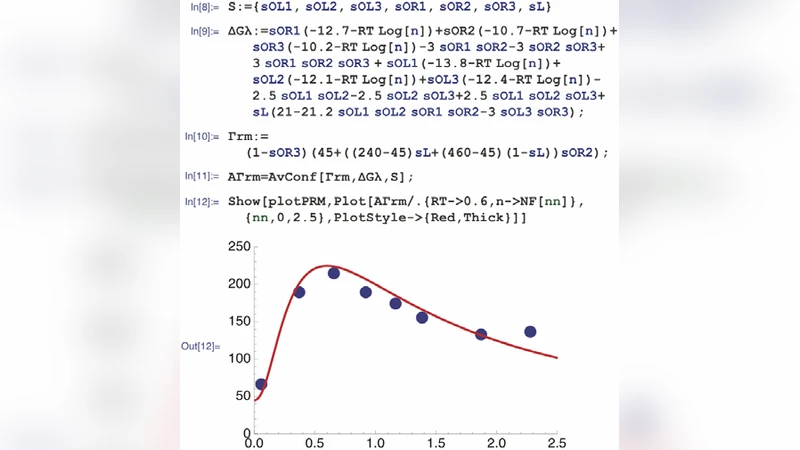
To demonstrate the utility of CplexA, the authors present three case studies. The first revisits the classic Escherichia coli lac operon, incorporating the LacI repressor, the CRP activator, and DNA looping between O1 and O3 operators. Using experimentally measured binding constants, CplexA reproduces the observed β‑galactosidase activity across a range of inducer concentrations and predicts the effect of mutating the looping segment. The second example tackles a mammalian multi‑enhancer promoter, such as the human β‑globin locus control region, where distal enhancers interact through chromatin loops. CplexA quantifies the cooperative contribution of each enhancer, matches DNase‑I footprinting data, and predicts how altering enhancer spacing changes transcriptional output. The third case integrates a MAPK‑dependent TF (e.g., ELK1) whose phosphorylation state modulates DNA affinity. By treating phosphorylation as a reversible modification with its own free‑energy term, the package simulates time‑dependent transcriptional responses to upstream signaling, illustrating how CplexA can bridge signal transduction and gene‑regulatory modeling.
The discussion acknowledges both strengths and limitations. CplexA’s main advantage is its exact treatment of the full thermodynamic ensemble without stochastic sampling, providing deterministic results that can be directly compared to quantitative experiments. Its integration with Mathematica’s plotting and notebook environment facilitates rapid prototyping and visual analysis. However, the approach relies on accurate thermodynamic parameters; uncertainties in ΔG values propagate to the predicted probabilities. Moreover, while the algorithm scales linearly with the number of interacting clusters, very large systems (hundreds of sites) may still strain memory resources. The authors propose future extensions such as sparse‑matrix representations, GPU‑accelerated convolution, and Bayesian inference modules to estimate parameter uncertainties from experimental data.
In conclusion, CplexA offers a powerful, user‑friendly platform for the quantitative analysis of complex gene‑regulatory architectures. By providing exact ensemble averages from a compact set of energetic inputs, it enables researchers to test mechanistic hypotheses, design synthetic promoters with desired logical behavior, and explore the impact of distal DNA contacts on transcriptional control. The package, together with comprehensive documentation and example notebooks, is freely available on SourceForge, positioning it as a valuable resource for systems biology, synthetic biology, and computational genomics communities.
Comments & Academic Discussion
Loading comments...
Leave a Comment