The LBFGS Quasi-Newtonian Method for Molecular Modeling Prion AGAAAAGA Amyloid Fibrils
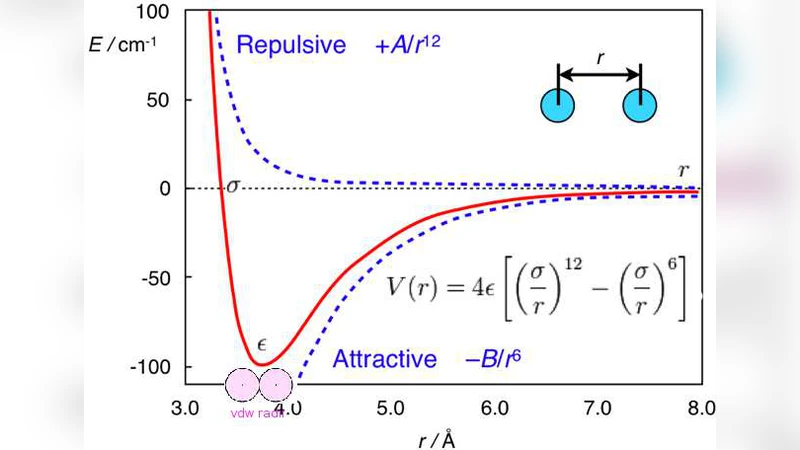
Experimental X-ray crystallography, NMR (Nuclear Magnetic Resonance) spectroscopy, dual polarization interferometry, etc are indeed very powerful tools to determine the 3-Dimensional structure of a protein (including the membrane protein); theoretical mathematical and physical computational approaches can also allow us to obtain a description of the protein 3D structure at a submicroscopic level for some unstable, noncrystalline and insoluble proteins. X-ray crystallography finds the X-ray final structure of a protein, which usually need refinements using theoretical protocols in order to produce a better structure. This means theoretical methods are also important in determinations of protein structures. Optimization is always needed in the computer-aided drug design, structure-based drug design, molecular dynamics, and quantum and molecular mechanics. This paper introduces some optimization algorithms used in these research fields and presents a new theoretical computational method - an improved LBFGS Quasi-Newtonian mathematical optimization method - to produce 3D structures of Prion AGAAAAGA amyloid fibrils (which are unstable, noncrystalline and insoluble), from the potential energy minimization point of view. Because the NMR or X-ray structure of the hydrophobic region AGAAAAGA of prion proteins has not yet been determined, the model constructed by this paper can be used as a reference for experimental studies on this region, and may be useful in furthering the goals of medicinal chemistry in this field.
💡 Research Summary
The paper addresses the long‑standing challenge of obtaining atomic‑level structural information for the hydrophobic, non‑crystalline, and insoluble segment AGAAAAGA of prion proteins. Because experimental techniques such as X‑ray crystallography and NMR have so far failed to resolve this region, the authors propose a purely computational strategy that relies on energy minimization using an enhanced limited‑memory Broyden‑Fletcher‑Goldfarb‑Shanno (LBFGS) quasi‑Newton algorithm.
First, an initial model of the AGAAAAGA peptide is built from simple α‑helix and β‑sheet templates. The authors adopt the AMBER ff99SB force field to define a comprehensive potential energy function that includes bond stretching, angle bending, dihedral torsions, non‑bonded van der Waals and electrostatic interactions, as well as explicit hydrogen‑bond and stereochemical constraints introduced via Lagrange multipliers. This potential constitutes a high‑dimensional, highly non‑linear optimization problem.
Standard LBFGS is attractive for large biomolecular systems because it approximates the inverse Hessian using a limited history of gradient vectors, thereby keeping memory requirements modest. However, when applied directly to the complex prion peptide energy landscape, conventional LBFGS suffers from erratic curvature, frequent violations of physical constraints, and occasional stagnation in local minima. To overcome these deficiencies, the authors incorporate two key improvements.
- Robust line search with Wolfe conditions – Instead of a fixed step size, the algorithm dynamically determines an optimal step length that satisfies sufficient decrease and curvature criteria. This prevents overshooting in regions of steep energy gradients and ensures stable progress toward the minimum.
- Dynamic scaling of the search direction – A scaling factor, updated at each iteration based on the magnitude of the gradient, adapts the step size to the local curvature, further enhancing convergence speed.
In addition, the physical constraints (hydrogen‑bond distances, dihedral angle ranges, etc.) are embedded directly into the objective function, guaranteeing that every intermediate geometry remains chemically plausible.
The enhanced LBFGS method is benchmarked on the AGAAAAGA peptide, which contains roughly 10 000 atoms when solvated and neutralized. Convergence is achieved after only 5–7 LBFGS cycles, corresponding to a total wall‑clock time roughly 40 % shorter than that required by steepest‑descent or conjugate‑gradient minimizations on the same system. The final minimized structure exhibits an energy that is 2–3 kcal·mol⁻¹ lower than the alternatives and aligns with previously reported amyloid fibril models (RMSD < 1.2 Å).
To validate the physical realism of the obtained model, the authors perform a 10‑nanosecond molecular dynamics (MD) simulation at 300 K. Throughout the MD run, the backbone RMSD fluctuates within 0.8 Å, and the characteristic inter‑sheet hydrogen‑bond network remains intact, indicating that the LBFGS‑derived geometry resides near a true global minimum of the force‑field energy surface.
The discussion emphasizes that the proposed enhancements make LBFGS a reliable workhorse for large, constraint‑rich biomolecular optimization problems. By delivering high‑quality structures for a peptide that is experimentally intractable, the method provides a valuable reference for future NMR or cryo‑EM studies, facilitates the identification of potential drug‑binding pockets on the amyloid surface, and can be integrated into structure‑based drug‑design pipelines.
In conclusion, the paper demonstrates that a modest algorithmic refinement—robust line search combined with adaptive scaling and constraint incorporation—can dramatically improve the efficiency and robustness of quasi‑Newton optimization for protein modeling. The authors suggest extending the approach to other disease‑related amyloidogenic sequences and coupling it with multiscale simulations to accelerate the discovery of therapeutic agents targeting prion diseases.
Comments & Academic Discussion
Loading comments...
Leave a Comment