A three-dimensional self-learning kinetic Monte Carlo model: application to Ag(111)
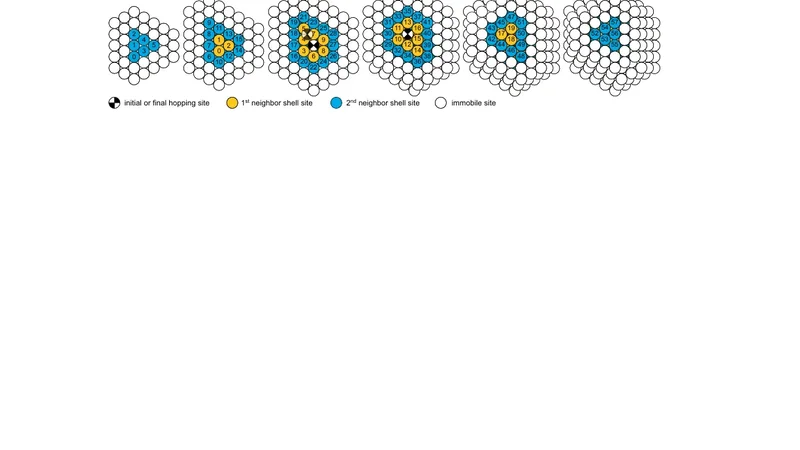
The reliability of kinetic Monte Carlo (KMC) simulations depends on accurate transition rates. The self-learning KMC method (Trushin et al 2005 Phys. Rev. B 72 115401) combines the accuracy of rates calculated from a realistic potential with the efficiency of a rate catalog, using a pattern recognition scheme. This work expands the original two-dimensional method to three dimensions. The concomitant huge increase in the number of rate calculations on the fly needed can be avoided by setting up an initial database, containing exact activation energies calculated for processes gathered from a simpler KMC model. To provide two representative examples, the model is applied to the diffusion of Ag monolayer islands on Ag(111), and the homoepitaxial growth of Ag on Ag(111) at low temperatures.
💡 Research Summary
The paper addresses a central challenge in kinetic Monte Carlo (KMC) simulations of atomistic processes: how to obtain accurate transition rates without prohibitive computational cost. Building on the self‑learning KMC (SL‑KMC) framework introduced by Trushin et al. (2005), which combines on‑the‑fly calculation of activation energies from a realistic interatomic potential with a pattern‑recognition based rate catalog, the authors extend the method from two‑dimensional (2D) surface diffusion to fully three‑dimensional (3D) systems.
In three dimensions the number of possible local atomic environments grows combinatorially, making a pure on‑the‑fly approach infeasible. To overcome this, the authors propose a two‑stage database construction strategy. First, a simplified KMC model—using only nearest‑neighbor interactions or a coarse‑grained energy rule—is employed to generate a large set of representative diffusion events on the Ag(111) surface. For each event the exact activation energy is computed once using an Embedded‑Atom Method (EAM) potential and stored in an initial catalog. Second, during the actual 3D SL‑KMC simulation, any newly encountered local configuration triggers an on‑the‑fly energy calculation, after which the result is added to the catalog for future reuse. This hybrid approach dramatically reduces the number of expensive energy evaluations while preserving the fidelity of the underlying physics.
The methodology is validated on two benchmark problems. (1) Diffusion of monolayer Ag islands on Ag(111). The authors show that island mobility depends sensitively on island size, shape, and the coordination of edge atoms. The 3D SL‑KMC captures not only edge atom detachment/attachment but also multi‑layer rearrangements that are invisible to 2D models. Consequently, the temperature dependence of the island diffusion coefficient matches experimental trends more closely than previous KMC studies. (2) Homoepitaxial growth of Ag at low temperatures (≈150 K). At such temperatures surface diffusion is strongly suppressed, leading to a competition between layer‑by‑layer growth and three‑dimensional nucleation. The 3D SL‑KMC reproduces the experimentally observed transition by accounting for cooperative multi‑atom moves that lower effective activation barriers. The simulations predict the critical flux and temperature at which the growth mode switches, providing insight into the role of collective atomic events that are often omitted in conventional KMC.
A systematic analysis of the initial catalog’s composition reveals that the choice of the simplified KMC model strongly influences overall efficiency. A well‑designed sampling scheme that covers a broad spectrum of local environments minimizes the frequency of on‑the‑fly calculations, thereby shortening simulation time without sacrificing accuracy. Conversely, an undersampled catalog leads to frequent new‑pattern detections and a steep rise in computational cost.
In summary, the authors demonstrate that three‑dimensional self‑learning KMC can deliver quantitatively reliable predictions for complex surface phenomena while keeping computational demands tractable. By pre‑computing a representative set of activation energies from a simple KMC precursor and augmenting it dynamically, the method bridges the gap between high‑accuracy atomistic potentials and the speed required for large‑scale simulations. The work opens the door for applying SL‑KMC to a wide range of materials—metals, semiconductors, oxides—and to processes such as thin‑film growth, catalytic reactions, and defect migration where three‑dimensional atomic rearrangements play a pivotal role.