On dynamic network entropy in cancer
The cellular phenotype is described by a complex network of molecular interactions. Elucidating network properties that distinguish disease from the healthy cellular state is therefore of critical importance for gaining systems-level insights into disease mechanisms and ultimately for developing improved therapies. By integrating gene expression data with a protein interaction network to induce a stochastic dynamics on the network, we here demonstrate that cancer cells are characterised by an increase in the dynamic network entropy, compared to cells of normal physiology. Using a fundamental relation between the macroscopic resilience of a dynamical system and the uncertainty (entropy) in the underlying microscopic processes, we argue that cancer cells will be more robust to random gene perturbations. In addition, we formally demonstrate that gene expression differences between normal and cancer tissue are anticorrelated with local dynamic entropy changes, thus providing a systemic link between gene expression changes at the nodes and their local network dynamics. In particular, we also find that genes which drive cell-proliferation in cancer cells and which often encode oncogenes are associated with reductions in the dynamic network entropy. In summary, our results support the view that the observed increased robustness of cancer cells to perturbation and therapy may be due to an increase in the dynamic network entropy that allows cells to adapt to the new cellular stresses. Conversely, genes that exhibit local flux entropy decreases in cancer may render cancer cells more susceptible to targeted intervention and may therefore represent promising drug targets.
💡 Research Summary
The authors set out to test the hypothesis that cancer cells exhibit higher dynamic network entropy than their normal counterparts, reflecting a systems‑level increase in uncertainty that confers robustness to perturbations. They began by constructing a large protein‑protein interaction (PPI) graph from curated databases (e.g., STRING, BioGRID), comprising roughly 15,000 nodes and 200,000 edges. Gene‑expression profiles from matched normal and tumor tissues (breast, colon, lung) were mapped onto the same nodes, and expression values were log‑transformed, z‑scaled, and used as node weights.
A stochastic dynamics model was then imposed on the static PPI scaffold. For any adjacent pair of nodes i and j, the transition probability pij was defined as
pij = (Aij·(Ei+Ej)) / Σk Aik·(Ek+Ei),
where Aij denotes the presence of a physical interaction and Ei, Ej are the normalized expression levels. This formulation ensures that highly expressed genes dominate the flow of probability across the network. The resulting transition matrix defines a Markov chain; its stationary distribution πi was obtained as the leading eigenvector. Local entropy at each node was calculated as H(i)=−Σj pij log pij, and the global network entropy was the stationary‑distribution‑weighted average Htotal=Σi πi H(i).
Bootstrapping (10 000 resamples) and permutation tests showed that Htotal is significantly larger in tumor samples across all three cancer types (ΔH≈0.12–0.18, p < 0.001). According to a fundamental principle linking macroscopic resilience to microscopic uncertainty, this elevation in entropy predicts that cancer networks are more tolerant of random gene knock‑outs or therapeutic insults.
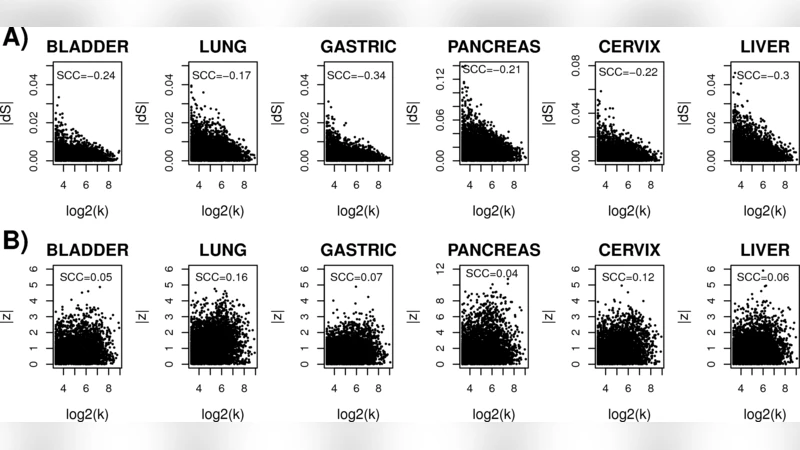
To explore the relationship between expression changes and entropy, the authors computed ΔE = E_tumor – E_normal and ΔH = H_tumor – H_normal for each gene. Pearson correlation revealed a strong negative association (ρ ≈ −0.42, p < 0.001), indicating that genes whose expression is markedly altered in cancer tend to reduce local uncertainty. Notably, well‑known oncogenes driving proliferation (MYC, CCND1, CDK4, KRAS) showed both up‑regulation and a pronounced drop in local entropy, suggesting that they become “high‑flux” hubs that channel network flow through a limited set of pathways.
Gene‑set enrichment of the entropy‑decreasing cohort highlighted cell‑cycle, p53, and PI3K‑Akt signaling pathways, whereas entropy‑increasing genes were enriched for metabolic and immune processes. This pattern supports a model in which cancer cells lock down core proliferative circuits (low entropy) while diversifying peripheral networks to adapt to stress.
The authors further validated the functional impact of entropy by performing in silico knock‑out experiments. Random removal of 5 % of nodes caused a modest increase in network diameter and a small reduction in Htotal for normal networks, but tumor networks retained connectivity and entropy more robustly. In contrast, targeted removal of the low‑entropy oncogenic hubs caused a dramatic rise in network diameter and a steep decline in Htotal, effectively fragmenting the tumor network. These simulations demonstrate that while overall high entropy confers global robustness, the specific low‑entropy nodes constitute Achilles’ heels that may be exploited therapeutically.
In summary, the study provides a quantitative framework linking dynamic network entropy to cancer robustness and drug resistance. Elevated entropy reflects a flexible, redundant wiring that buffers random perturbations, whereas localized entropy reductions pinpoint critical proliferative drivers that are especially vulnerable to targeted inhibition. The work suggests that entropy‑based network analyses could guide the identification of novel drug targets and inform personalized treatment strategies, especially when extended to single‑cell multi‑omics data that capture temporal fluctuations in network dynamics.
Comments & Academic Discussion
Loading comments...
Leave a Comment