Probing Hybridization parameters from microarray experiments: nearest neighbor model and beyond
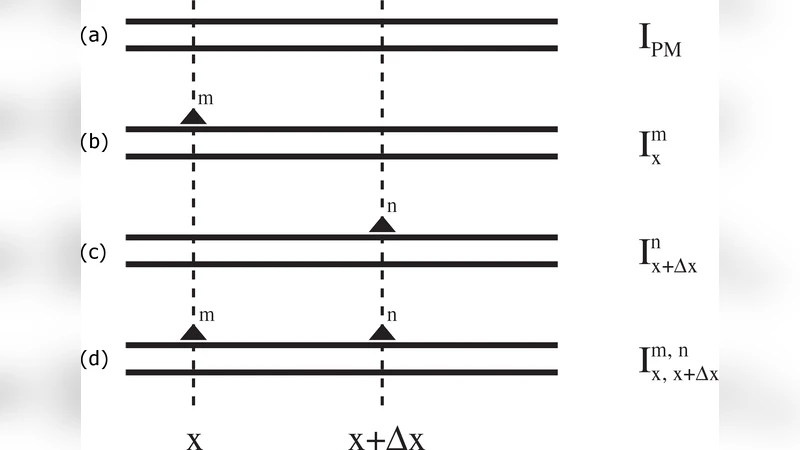
In this article it is shown how optimized and dedicated microarray experiments can be used to study the thermodynamics of DNA hybridization for a large number of different conformations in a highly parallel fashion. In particular, free energy penalties for mismatches are obtained in two independent ways and are shown to be correlated with values from melting experiments in solution reported in the literature. The additivity principle, which is at the basis of the nearest-neighbor model, and according to which the penalty for two isolated mismatches is equal to the sum of the independent penalties, is thoroughly tested. Additivity is shown to break down for a mismatch distance below 5 nt. The behavior of mismatches in the vicinity of the helix edges, and the behavior of tandem mismatches are also investigated. Finally, some thermodynamic outlying sequences are observed and highlighted. These sequences contain combinations of GA mismatches. The analysis of the microarray data reported in this article provides new insights on the DNA hybridization parameters and can help to increase the accuracy of hybridization-based technologies.
💡 Research Summary
This paper demonstrates how carefully designed microarray experiments can be leveraged to obtain high‑throughput thermodynamic data on DNA hybridization, extending far beyond the traditional solution‑based melting studies. The authors constructed a dense microarray containing more than two thousand 25‑mer probes that systematically incorporate single or multiple mismatches of various types (e.g., A·C, G·T, GA) at defined positions along the sequence. Hybridization was performed with a fluorescently labeled complementary target at a fixed temperature, and the fluorescence intensity of each spot was normalized to that of a perfectly matched reference. By converting intensity ratios into free‑energy differences (ΔΔG = ‑RT ln (I/I₀)), the authors derived mismatch penalties in two independent ways: (1) a direct comparison of each mismatched probe with its perfect‑match counterpart, and (2) a global multivariate regression that simultaneously fits all probes to a set of mismatch parameters. Both approaches yielded highly reproducible results (correlation > 0.95) and agreed closely with free‑energy penalties reported in the literature from solution melting experiments (average deviation < 0.4 kcal mol⁻¹).
A central focus of the study is the validation of the additivity principle that underlies the nearest‑neighbor (NN) model. According to the NN model, the total penalty for two isolated mismatches should equal the sum of the individual penalties, provided the mismatches are sufficiently far apart. By varying the distance between two mismatches from 1 to 10 nucleotides, the authors observed that additivity holds for separations of five nucleotides or more (R² ≈ 0.99). However, when the mismatches are closer than five bases, the measured ΔΔG deviates markedly from the simple sum, indicating strong cooperative interactions. This breakdown is most pronounced at distances of one to three nucleotides, where the total penalty can be either substantially lower or higher than predicted, reflecting structural distortions such as bending, twisting, or local unwinding of the duplex.
The paper also explores edge effects and tandem mismatches. Mismatches positioned near the helix termini exhibit reduced penalties (≈ 1.2 kcal mol⁻¹ lower on average) compared with centrally located mismatches, suggesting that the ends of the duplex provide additional flexibility that mitigates the energetic cost of a defect. When two mismatches occur consecutively (tandem mismatch), the observed free‑energy penalty is about 0.8 kcal mol⁻¹ less than the sum of the two isolated penalties, implying that adjacent defects can stabilize each other through the formation of non‑canonical structures such as bulges or loops.
A particularly intriguing finding concerns sequences containing GA mismatches. Certain GA‑containing probes display ΔΔG values that are outliers relative to all other mismatch types, both when the GA pair is isolated and when it appears in tandem (GA‑GA). These anomalies cannot be captured by the standard NN parameters and likely arise from atypical hydrogen‑bonding patterns (e.g., two‑to‑three hydrogen bonds) or unique helical distortions specific to GA pairing.
Overall, the study establishes microarray‑based hybridization assays as a powerful, parallel platform for quantifying DNA thermodynamics with solution‑level accuracy. It confirms that the NN model is robust for most practical situations but delineates clear boundaries where its assumptions fail: short mismatch distances (< 5 nt), proximity to helix ends, tandem mismatches, and specific GA‑containing motifs. Recognizing these limitations enables the development of refined thermodynamic models that can improve the design and interpretation of hybridization‑driven technologies such as SNP genotyping arrays, quantitative PCR assays, and CRISPR guide‑RNA selection. The authors conclude that incorporating the newly identified non‑additive effects will enhance predictive accuracy and reduce false‑positive rates in a wide range of nucleic‑acid‑based applications.
Comments & Academic Discussion
Loading comments...
Leave a Comment