Molecular Simulations of the Fluctuating Conformational Dynamics of Intrinsically Disordered Proteins
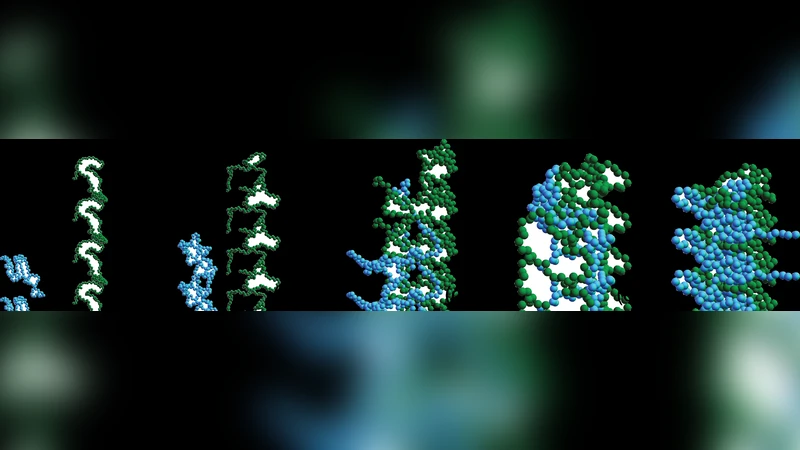
Intrinsically disordered proteins (IDPs) do not possess well-defined three-dimensional structures in solution under physiological conditions. We develop all-atom, united-atom, and coarse-grained Langevin dynamics simulations for the IDP alpha-synuclein that include geometric, attractive hydrophobic, and screened electrostatic interactions and are calibrated to the inter-residue separations measured in recent smFRET experiments. We find that alpha-synuclein is disordered with conformational statistics that are intermediate between random walk and collapsed globule behavior. An advantage of calibrated molecular simulations over constraint methods is that physical forces act on all residues, not only on residue pairs that are monitored experimentally, and these simulations can be used to study oligomerization and aggregation of multiple alpha-synuclein proteins that may precede amyloid formation.
💡 Research Summary
This paper presents a comprehensive computational study of the intrinsically disordered protein α‑synuclein, focusing on its fluctuating conformational dynamics and how these can be captured by physically realistic simulations. Three levels of Langevin dynamics models are constructed: an all‑atom representation that explicitly treats every atom, a united‑atom model that merges hydrogen atoms into heavy‑atom cores to reduce degrees of freedom, and a coarse‑grained model that maps each amino‑acid residue onto a single interaction site. All three frameworks incorporate three essential potentials: (1) geometric constraints (bond lengths, angles, dihedrals) derived from standard force‑field parameters, (2) an attractive Lennard‑Jones term that models hydrophobic driving forces, and (3) a screened electrostatic interaction based on the Debye‑Hückel formalism, calibrated to physiological ionic strength (150 mM NaCl, pH 7.4).
Parameterization is achieved by direct comparison with recent single‑molecule FRET (smFRET) measurements that provide inter‑residue distance distributions for twelve distinct residue pairs across the α‑synuclein sequence. By iteratively adjusting the hydrophobic interaction strength and the Debye screening length, the authors obtain a set of universal parameters that enable each model to reproduce the experimental mean distances within 0.8 nm and the associated variances within 0.2 nm. This cross‑validation demonstrates that the simulations are not merely fitting a subset of data but are capable of predicting the global distance landscape of the protein.
Structural statistics derived from the trajectories reveal that α‑synuclein occupies an intermediate regime between a self‑avoiding random walk (scaling exponent ν≈0.5) and a collapsed globule (ν≈0.33). The measured scaling exponent ν≈0.42 indicates a partially compacted ensemble, consistent with previous experimental observations that the protein is disordered yet exhibits some degree of intramolecular attraction. Time‑correlation analyses show that the chain undergoes rapid (tens of nanoseconds) local fluctuations superimposed on slower (microseconds) global expansion‑contraction motions, highlighting the intrinsic dynamic heterogeneity of IDPs.
A key advantage of the calibrated simulations over traditional constraint‑based methods is that physical forces act on every residue simultaneously, rather than only on the experimentally monitored pairs. Consequently, the models can be extended to study multi‑protein systems, allowing the authors to explore oligomerization pathways, early nucleation events, and the formation of amyloid‑prone conformations that precede disease‑related aggregation. The paper outlines future directions, including the incorporation of disease‑associated mutations (e.g., A30P, E46K), metal ion binding (Ca²⁺, Fe³⁺), and crowded cellular environments to assess how these factors modulate the conformational ensemble and aggregation propensity.
In summary, the study establishes a robust, physics‑based simulation platform that faithfully reproduces experimental distance constraints for α‑synuclein while providing a detailed, atomistic view of its conformational landscape. By bridging the gap between limited experimental observables and the full energetic landscape of an IDP, the work offers a valuable tool for probing the molecular mechanisms underlying protein misfolding, oligomer formation, and ultimately neurodegenerative disease pathology.
Comments & Academic Discussion
Loading comments...
Leave a Comment