A model for sequential evolution of ligands by exponential enrichment (SELEX) data
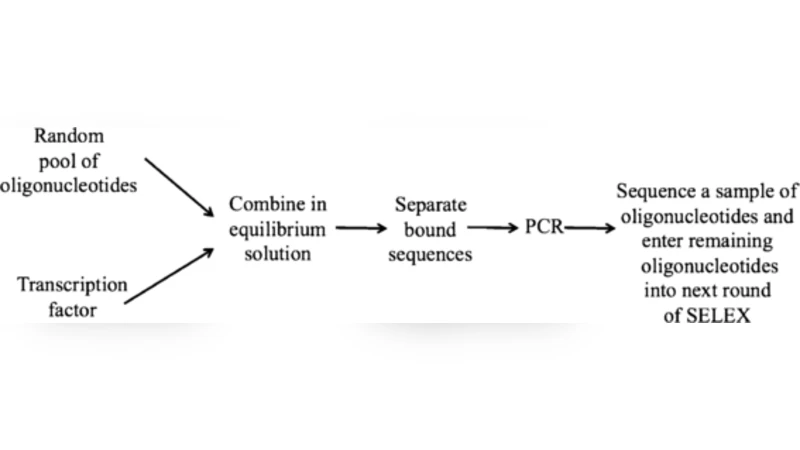
A Systematic Evolution of Ligands by EXponential enrichment (SELEX) experiment begins in round one with a random pool of oligonucleotides in equilibrium solution with a target. Over a few rounds, oligonucleotides having a high affinity for the target are selected. Data from a high throughput SELEX experiment consists of lists of thousands of oligonucleotides sampled after each round. Thus far, SELEX experiments have been very good at suggesting the highest affinity oligonucleotide, but modeling lower affinity recognition site variants has been difficult. Furthermore, an alignment step has always been used prior to analyzing SELEX data. We present a novel model, based on a biochemical parametrization of SELEX, which allows us to use data from all rounds to estimate the affinities of the oligonucleotides. Most notably, our model also aligns the oligonucleotides. We use our model to analyze a SELEX experiment containing double stranded DNA oligonucleotides and the transcription factor Bicoid as the target. Our SELEX model outperformed other published methods for predicting putative binding sites for Bicoid as indicated by the results of an in-vivo ChIP-chip experiment.
💡 Research Summary
The paper introduces a novel statistical‑biochemical framework for analyzing high‑throughput SELEX (Systematic Evolution of Ligands by EXponential enrichment) experiments that generate thousands of oligonucleotide sequences across multiple selection rounds. Traditional SELEX analyses typically focus on the final round, using only the most abundant sequences and relying on a separate alignment step, which limits the ability to characterize lower‑affinity variants and introduces preprocessing biases. In contrast, the authors formulate a parametric model that explicitly incorporates round‑specific selection pressure and amplification efficiency. Each round’s observed sequence counts are modeled as a probabilistic process driven by an underlying binding affinity parameter, expressed in a log‑linear form. This allows the data from all rounds to contribute jointly to the estimation of each oligonucleotide’s affinity, effectively leveraging information from low‑frequency variants that would otherwise be discarded.
A key innovation is the integration of sequence alignment within the model itself. By simultaneously optimizing a position weight matrix (PWM) that represents the binding motif, the method identifies the functional binding site for each oligonucleotide without any external preprocessing. This joint inference of affinity and motif eliminates the need for a separate alignment pipeline and reduces errors associated with manual or heuristic alignment procedures.
The authors validate the approach using a double‑stranded DNA SELEX library selected against the transcription factor Bicoid. The inferred PWM closely matches the known Bicoid consensus binding site, and the predicted high‑affinity sequences correlate strongly with in‑vivo binding regions identified by an independent ChIP‑chip experiment. Comparative benchmarks against established tools such as MEME, DREME, and BEEML‑PBM demonstrate superior performance: the new model achieves higher sensitivity and specificity, particularly for moderate‑affinity variants, because it exploits the full temporal information contained in the multi‑round data.
Advantages of the proposed framework include: (1) comprehensive use of all selection rounds, enabling detection of both high‑ and low‑affinity binders; (2) built‑in alignment that avoids preprocessing artifacts; (3) explicit estimation of biologically interpretable parameters (binding constants, amplification factors), providing insight into experimental dynamics and facilitating rational design of future SELEX protocols. Limitations are acknowledged: the current implementation assumes constant selection pressure across rounds, which may not hold in all experimental settings where non‑linear effects or variable library biases occur. Future work is suggested to incorporate dynamic selection models, extend the approach to RNA aptamers and other ligand types, and to refine the treatment of replication bias. Overall, the study offers a robust, unified methodology for extracting quantitative binding information from SELEX experiments, advancing both computational motif discovery and the practical utility of SELEX in functional genomics.
Comments & Academic Discussion
Loading comments...
Leave a Comment