Correlation Energy Divergences in Metallic Systems
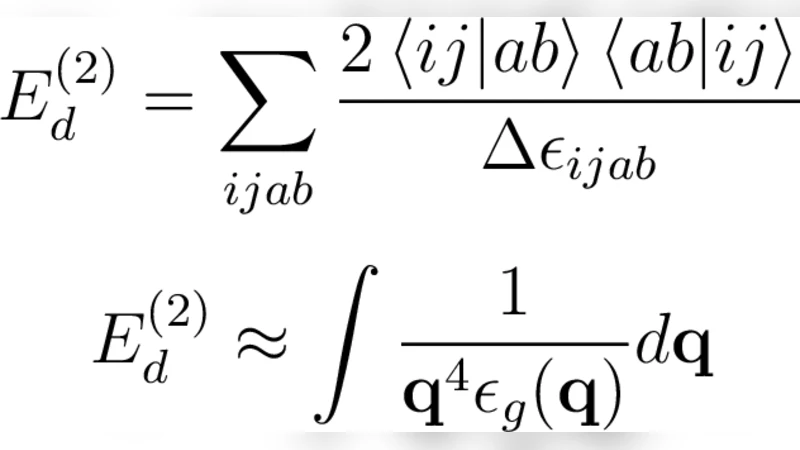
We numerically examine divergences of the total energy in metallic systems of approximate many-body theories using Hartree–Fock as a reference, including perturbative (M\oller-Plesset, MP), coupled cluster (CC) and configuration interaction (CI) approaches. Controlling for finite size effects and basis set incompleteness error by comparison with energies from the random phase approximation (RPA), we suggest convincingly that non-perturbative coupled cluster theories are acceptable for modelling electronic interactions in metals whilst perturbative coupled cluster theories are not. Data are provided from the RPA with which it is possible to test other approximate finite-basis methods for divergences with only modest computational cost.
💡 Research Summary
This paper investigates the long‑standing problem of correlation‑energy divergences in metallic systems when using approximate many‑body electronic‑structure methods that are based on a Hartree–Fock (HF) reference. Metals, modeled here as a three‑dimensional homogeneous electron gas, possess a continuous Fermi surface; consequently, perturbative expansions that contain energy denominators approaching zero can produce unphysical divergences as the system size grows. The authors systematically benchmark a suite of methods—second‑order Møller–Plesset perturbation theory (MP2), third‑order MP3, configuration interaction with single and double excitations (CISD), coupled‑cluster singles and doubles (CCSD), and the perturbative triples correction CCSD(T)—against the Random Phase Approximation (RPA). RPA is chosen as a reference because it treats long‑range electron–electron screening exactly and yields a well‑behaved energy in the thermodynamic limit, thus providing a reliable yardstick for both finite‑size and basis‑set errors.
Methodologically, the study varies the number of electrons (N) and the size of the Gaussian basis set (M) to isolate finite‑size effects from basis‑set incompleteness. For each combination, total energies are computed with the aforementioned methods and compared to the corresponding RPA energy. The results demonstrate a clear dichotomy: MP2 and MP3 energies drift toward negative infinity as N increases, confirming the expected divergence of low‑order perturbation theory in a metal. CISD shows a similar trend, albeit with a slower divergence due to its limited excitation space. In stark contrast, CCSD and especially CCSD(T) produce energies that converge smoothly to the RPA value, indicating that the non‑perturbative resummation inherent in coupled‑cluster theory successfully regularizes the problematic denominators. The inclusion of perturbative triples in CCSD(T) further improves agreement with RPA without incurring prohibitive computational cost, suggesting that CCSD(T) is a practical, high‑accuracy tool for metallic systems.
The authors discuss why coupled‑cluster methods succeed where perturbative approaches fail. By iteratively solving amplitude equations, CCSD effectively sums an infinite subset of diagrams, thereby capturing screening and collective excitations that are essential in a metal’s continuous spectrum. The perturbative triples correction adds a controlled, higher‑order refinement that does not re‑introduce divergence. The paper also emphasizes the utility of the supplied RPA dataset: researchers can test new approximate methods (e.g., DFT+U, GW, DMFT, or machine‑learned functionals) for divergence behavior at modest computational expense.
In conclusion, the study provides compelling numerical evidence that non‑perturbative coupled‑cluster theories, particularly CCSD and CCSD(T), are robust and accurate for modeling electronic interactions in metals, whereas low‑order perturbative schemes such as MP2 are intrinsically unsuitable due to their divergent nature. The work paves the way for broader adoption of coupled‑cluster techniques in solid‑state physics and materials science, and it highlights the importance of benchmark references like RPA for validating emerging electronic‑structure approximations.