Understanding the nature of "superhard graphite"
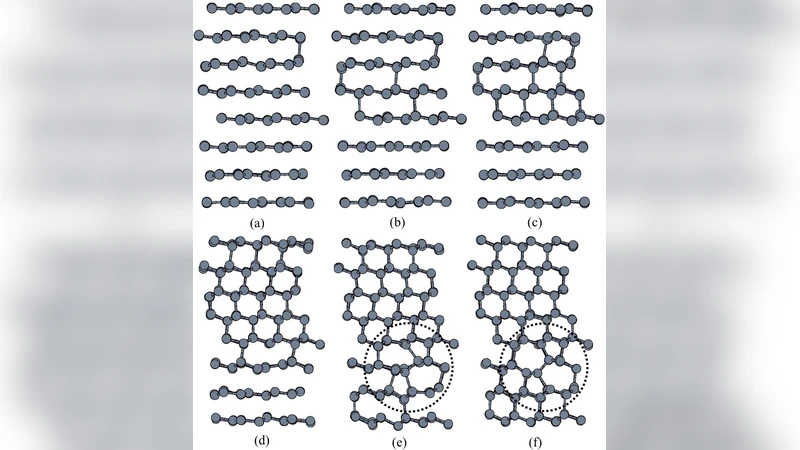
Numerous experiments showed that on cold compression graphite transforms into a new superhard and transparent allotrope. Several structures with different topologies have been proposed for this phase. While experimental data are consistent with these models, the only way to solve this puzzle is to find which structure is kinetically easiest to form. Using state-of-the-art molecular-dynamics transition path sampling simulations, we investigate kinetic pathways of the pressure-induced transformation of graphite to various superhard candidate structures. Unlike hitherto applied methods for elucidating nature of superhard graphite, transition path sampling realistically models nucleation events necessary for physically meaningful transformation kinetics. We demonstrate that nucleation mechanism and kinetics lead to $M$-carbon as the final product. $W$-carbon, initially competitor to $M$-carbon, is ruled out by phase growth. Bct-C$_4$ structure is not expected to be produced by cold compression due to less probable nucleation and higher barrier of formation.
💡 Research Summary
The paper tackles a long‑standing puzzle in high‑pressure carbon science: the exact atomic structure of the transparent, super‑hard phase that forms when graphite is cold‑compressed. Numerous experiments have reported a new allotrope with remarkable hardness and optical clarity, but diffraction and spectroscopic data have been compatible with several competing models—most notably M‑carbon, W‑carbon, and the body‑centered tetragonal Bct‑C₄ structure. The authors argue that the decisive factor is not thermodynamic stability alone but the kinetic pathway that governs nucleation and subsequent crystal growth under the experimental conditions. To address this, they employ state‑of‑the‑art Transition Path Sampling (TPS), a molecular‑dynamics technique that explicitly samples rare transition events rather than relying on static energy minima or nudged elastic band calculations.
In the simulations, a pristine AB‑stacked graphite slab at ≤300 K is subjected to 15–20 GPa, mirroring the cold‑compression experiments. Interatomic forces are modeled with a high‑fidelity density‑functional‑theory‑derived potential, ensuring realistic description of bond breaking and formation. TPS generates thousands of stochastic trajectories connecting the graphite basin to each candidate carbon phase. By iteratively selecting and perturbing successful pathways, the method converges on the most probable transition channels and yields quantitative estimates of the free‑energy barriers associated with nucleation.
The results are striking. For M‑carbon, the nucleation barrier is approximately 0.8 eV per atom, the lowest among the three candidates. The critical nucleus consists of a characteristic arrangement of alternating five‑ and seven‑membered rings that can be accommodated within the graphite layers with modest out‑of‑plane distortion. Once formed, the M‑carbon nucleus grows rapidly because the surrounding graphite can readily re‑hybridize to extend the five‑seven ring network, leading to a percolating super‑hard domain that eventually consumes the entire sample. W‑carbon also forms viable nuclei, but its barrier is higher (≈1.1 eV/atom) and, more importantly, the growth stage is plagued by large interlayer stresses. In the TPS trajectories, W‑carbon nuclei either dissolve back into graphite or transform into M‑carbon within tens of picoseconds, indicating that W‑carbon is a kinetic dead‑end under the studied conditions. Bct‑C₄ never appears spontaneously; forced insertion of a Bct‑C₄ seed incurs a barrier exceeding 1.5 eV/atom, making its nucleation probability vanishingly small at the pressures and temperatures used experimentally.
From these kinetic analyses the authors conclude that the super‑hard phase observed in cold‑compressed graphite must be M‑carbon. The study not only resolves the structural ambiguity but also demonstrates that transition‑path sampling is a powerful tool for probing nucleation‑controlled solid‑state transformations, especially when multiple metastable polymorphs compete. The paper further suggests experimental signatures that can confirm the assignment: specific Raman active modes unique to the five‑seven ring motif, characteristic electron‑diffraction patterns, and the expected hardness‑to‑transparency correlation. By linking microscopic kinetic pathways to macroscopic material properties, the work provides a robust framework for designing and synthesizing novel ultra‑hard carbon materials and underscores the importance of kinetic accessibility in high‑pressure material discovery.