Efficient Prediction of DNA-Binding Proteins Using Machine Learning
DNA-binding proteins are a class of proteins which have a specific or general affinity to DNA and include three important components: transcription factors; nucleases, and histones. DNA-binding proteins also perform important roles in many types of cellular activities. In this paper we describe machine learning systems for the prediction of DNA- binding proteins where a Support Vector Machine and a Cascade Correlation Neural Network are optimized and then compared to determine the learning algorithm that achieves the best prediction performance. The information used for classification is derived from characteristics that include overall charge, patch size and amino acids composition. In total 121 DNA- binding proteins and 238 non-binding proteins are used to build and evaluate the system. For SVM using the ANOVA Kernel with Jack-knife evaluation, an accuracy of 86.7% has been achieved with 91.1% for sensitivity and 85.3% for specificity. For CCNN optimized over the entire dataset with Jack knife evaluation we report an accuracy of 75.4%, while the values of specificity and sensitivity achieved were 72.3% and 82.6%, respectively.
💡 Research Summary
The paper addresses the computational prediction of DNA‑binding proteins (DNA‑BPs), a task of considerable biological importance because DNA‑BPs such as transcription factors, nucleases, and histones regulate a wide array of cellular processes. Experimental identification of DNA‑BPs is labor‑intensive and costly, motivating the development of in‑silico methods that can rapidly screen protein sequences for DNA‑binding propensity.
Dataset
A curated benchmark comprising 121 experimentally verified DNA‑binding proteins and 238 non‑binding proteins was assembled from public repositories and prior literature. The authors took care to remove redundancy and to balance the classes, though the non‑binding set remains larger to mitigate class‑imbalance effects.
Feature Engineering
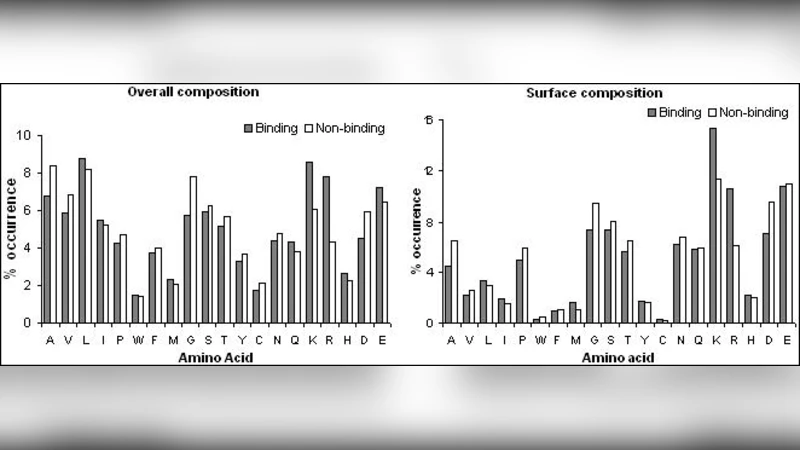
Three physicochemical descriptors were extracted for each protein: (1) overall net charge, reflecting electrostatic attraction to the negatively charged DNA backbone; (2) patch size, defined as the surface area of contiguous charged residues that could form a binding interface; and (3) amino‑acid composition, i.e., the relative frequencies of the 20 standard residues, which captures sequence‑level preferences for DNA interaction. All features were normalized to zero mean and unit variance before feeding them to the classifiers.
Learning Algorithms
Two distinct machine learning paradigms were evaluated:
-
Support Vector Machine (SVM) – The authors selected the ANOVA kernel, a hybrid of polynomial and Gaussian kernels that can model complex, non‑linear relationships while controlling the dimensionality explosion typical of high‑order polynomial kernels. Hyper‑parameters (regularization parameter C and kernel width γ) were tuned via exhaustive grid search combined with cross‑validation.
-
Cascade Correlation Neural Network (CCNN) – CCNNs start with a minimal network and iteratively add hidden units that maximally correlate with the residual error of the current model. This dynamic growth can, in principle, adapt model capacity to the data. The authors trained the CCNN on the entire dataset, allowing the architecture to expand until performance plateaued, and they adjusted learning rates and regularization terms to avoid over‑fitting.
Evaluation Protocol
Because the dataset is modest in size, the authors employed a jack‑knife (leave‑one‑out) validation scheme. Each protein is held out once as a test case while the remaining samples train the model, ensuring that every observation contributes to both training and testing. Performance was quantified using accuracy, sensitivity (recall), and specificity.
Results
- SVM with ANOVA kernel achieved an overall accuracy of 86.7 %, a sensitivity of 91.1 %, and a specificity of 85.3 %.
- CCNN yielded an accuracy of 75.4 %, sensitivity of 82.6 %, and specificity of 72.3 %.
The SVM clearly outperformed the CCNN across all metrics, especially in sensitivity, indicating a superior ability to correctly identify true DNA‑binding proteins. The CCNN’s lower specificity suggests a higher false‑positive rate, which could be problematic in downstream experimental validation.
Discussion
The superiority of the SVM can be attributed to several factors. First, the ANOVA kernel efficiently captures the non‑linear interactions among the three engineered features without requiring a large number of parameters. Second, the rigorous jack‑knife validation reduces the risk of optimistic bias that can arise with small datasets. In contrast, the CCNN, while theoretically flexible, suffers from a relatively large parameter space relative to the limited training data, leading to over‑fitting and sub‑optimal generalization.
The feature set, though biologically motivated, is modest. It omits structural information such as three‑dimensional contact maps, electrostatic potential surfaces, and evolutionary conservation scores—all of which have been shown to improve DNA‑BP prediction in later studies. Consequently, the reported accuracies represent a baseline that could be substantially raised by integrating richer descriptors.
Limitations and Future Work
- Feature Expansion: Incorporating structural and evolutionary features (e.g., position‑specific scoring matrices, solvent accessibility, and DNA‑binding motifs) is expected to boost discriminative power.
- Data Scaling: As more experimentally validated DNA‑BPs become available, larger training sets will enable the use of deeper neural architectures and more robust statistical estimates.
- Ensemble Strategies: Combining the strengths of kernel‑based methods (high precision) with those of neural networks (capacity to learn hierarchical patterns) via ensemble or hybrid models could yield higher overall performance.
- Application‑Oriented Thresholding: In practical pipelines, adjusting decision thresholds to prioritize either sensitivity (for discovery) or specificity (for validation) may be beneficial; the current study provides a solid baseline for such tuning.
Conclusion
The study demonstrates that a carefully tuned SVM with an ANOVA kernel can achieve high‑quality prediction of DNA‑binding proteins using only three simple physicochemical descriptors, outperforming a dynamically grown cascade‑correlation neural network on the same data. The work establishes a benchmark for future methodological improvements and underscores the importance of feature selection, kernel choice, and rigorous validation in bioinformatics classification tasks.
Comments & Academic Discussion
Loading comments...
Leave a Comment