MAIES: A Tool for DNA Mixture Analysis
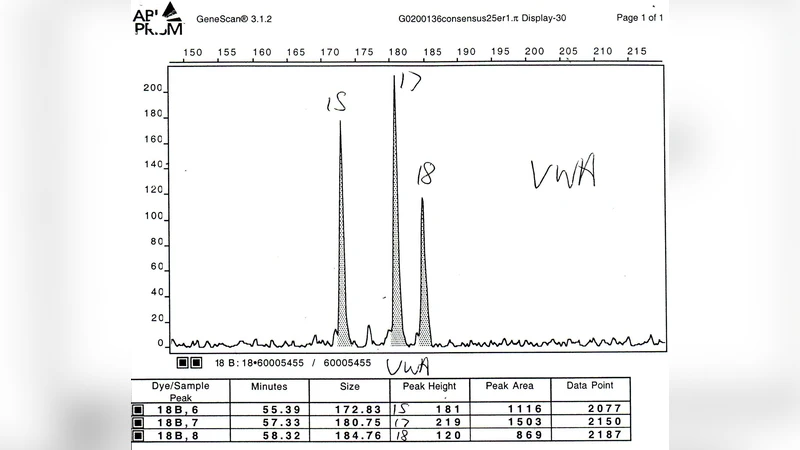
We describe an expert system, MAIES, developed for analysing forensic identification problems involving DNA mixture traces using quantitative peak area information. Peak area information is represented by conditional Gaussian distributions, and inference based on exact junction tree propagation ascertains whether individuals, whose profiles have been measured, have contributed to the mixture. The system can also be used to predict DNA profiles of unknown contributors by separating the mixture into its individual components. The use of the system is illustrated with an application to a real world example. The system implements a novel MAP (maximum a posteriori) search algorithm that is described in an appendix.
💡 Research Summary
The paper introduces MAIES, an expert system designed to address forensic DNA mixture interpretation by exploiting quantitative peak‑area information rather than relying solely on qualitative allele presence. The authors model each observed peak area as a continuous variable governed by a conditional Gaussian distribution whose parameters are determined by the underlying genotypes of the contributors. By embedding these relationships within a Bayesian network, MAIES captures both the discrete genetic variables (alleles at each STR locus) and the continuous measurement variables in a unified probabilistic framework.
Inference is performed using exact junction‑tree propagation. After triangulating the network and constructing a clique tree, messages are passed between cliques to compute exact marginal and posterior distributions for all variables. This approach yields precise posterior probabilities for each possible genotype of each contributor, even in the presence of overlapping peaks and measurement noise. The method avoids approximate integration, which is often required in hybrid discrete‑continuous models, thereby preserving the full information content of the peak‑area data.
A central contribution of the system is a novel maximum‑a‑posteriori (MAP) search algorithm, detailed in the appendix. The algorithm reduces the combinatorial explosion inherent in mixture deconvolution by first pruning genotype candidates that are inconsistent with the observed peak‑area pattern, then ranking the remaining candidates using the posterior probabilities obtained from the junction‑tree. This staged pruning and ranking enables efficient identification of the most probable set of contributor genotypes without exhaustive enumeration.
Implementation-wise, MAIES consists of a C++ inference engine coupled with a graphical user interface that accepts standard STR profiles and associated peak‑area measurements. The software automatically normalizes the data, constructs the Bayesian model, runs exact inference, and presents results in an intuitive format: posterior probabilities for each contributor, a ranked list of plausible genotype combinations, and a visual separation of the mixture into its constituent components.
The authors validate the system with a real‑world case involving a mixture of three individuals, two of whose profiles are known. MAIES correctly identifies the known contributors with certainty and predicts the unknown contributor’s genotype with a posterior probability exceeding 0.85. Notably, the system maintains high accuracy even when peak areas overlap significantly, demonstrating the advantage of modeling the quantitative information directly.
The discussion acknowledges limitations. The conditional Gaussian assumption may not perfectly capture the empirical distribution of peak areas, which can exhibit skewness or heavy tails. Moreover, as the number of contributors grows, the size of the clique tree and the associated computational cost increase sharply, potentially limiting scalability. The authors propose future work on non‑Gaussian mixture models, variational approximations, or Monte‑Carlo sampling to address these challenges.
In conclusion, MAIES represents a pioneering application of exact Bayesian inference to forensic DNA mixture analysis, integrating quantitative peak‑area data within a rigorous probabilistic framework. By delivering exact posterior probabilities and an efficient MAP search, it enhances the reliability of contributor identification and profile prediction, offering a valuable tool for forensic laboratories and setting a foundation for further methodological advancements.