Equilibrium insertion of nanoscale objects into phospholipid bilayers
Certain membrane proteins, peptides, nanoparticles and nanotubes have rigid structure and fixed shape. They are often viewed as spheres and cylinders with certain surface properties. Single Chain Mean Field theory is used to model the equilibrium insertion of nanoscale spheres and rods into the phospholipid bilayer. The equilibrium structures and the resulting free energies of the nano-objects in the bilayer allow to distinguish different orientations in the bilayer and estimate the energy barrier of insertion.
💡 Research Summary
**
This paper presents a quantitative theoretical study of how rigid nanoscale objects—specifically spheres and cylinders—insert into a phospholipid bilayer at equilibrium. The authors employ the Single Chain Mean Field (SCMF) approach, a self‑consistent field theory that treats each lipid molecule as a coarse‑grained three‑bead construct: two hydrophobic “T” beads and one hydrophilic “H” bead, all of equal radius (4.05 Å) and linked by rigid bonds of 10 Å. Pairwise interactions are modeled with short‑range attractive potentials (ε_TT = −2.10 kT for T‑T contacts, ε_HS = −0.15 kT for H‑solvent contacts). The total free energy comprises entropy contributions of lipids and solvent, intra‑molecular energy, inter‑bead interactions, and a steric incompressibility term enforced by a Lagrange multiplier.
Equilibrium is obtained by minimizing this free energy with respect to the single‑molecule probability distribution ρ(γ) and the solvent density c_S(r), leading to a set of coupled integral equations (Eqs. 6‑8). In practice the authors discretize space into 2 Å cells, sample 750 000 lipid conformations using the Rosenbluth algorithm, and replace integrals by finite sums. This sampling yields free‑energy estimates with an uncertainty of roughly ±10 kT.
Nanoparticles are introduced as excluded volumes that lipids and solvent cannot occupy; their surfaces can carry additional hydrophilic or hydrophobic interactions. Three geometries are examined: (i) a sphere, (ii) a cylinder oriented perpendicular to the bilayer (the “vertical” case), and (iii) a cylinder oriented parallel to the bilayer plane (the “parallel” case). For the sphere and vertical cylinder, a two‑dimensional cylindrical symmetry is used; for the parallel cylinder, an infinite‑cylinder approximation is adopted, with hard walls at the top and bottom of the simulation box to prevent the bilayer from escaping.
The main findings are as follows:
-
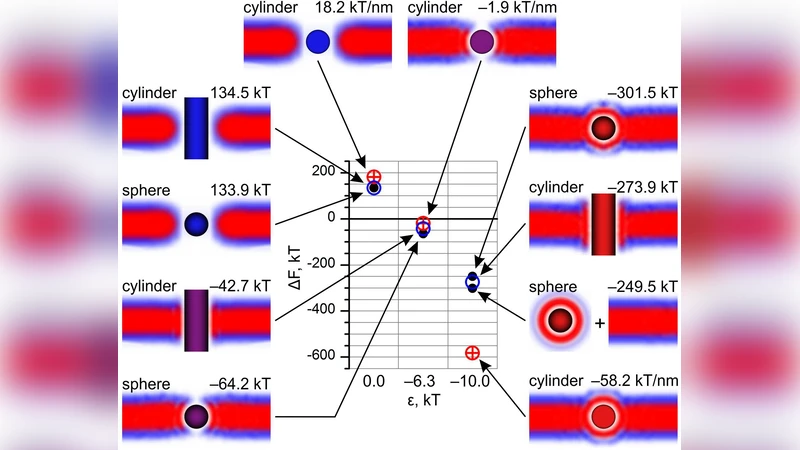
Spherical particles: When the sphere’s surface is predominantly hydrophobic, the free‑energy minimum occurs when the particle is centered within the hydrophobic core of the bilayer. The insertion barrier is modest, about 15 kT, consistent with experimental observations of membrane permeation for hydrophobic nanoparticles. Increasing the hydrophilic fraction raises the barrier sharply, indicating that surface chemistry critically controls insertion propensity.
-
Vertical cylinders: If the cylinder length matches the bilayer thickness (≈30 Å), the hydrophobic core of the cylinder aligns with the bilayer’s hydrophobic region, achieving optimal “hydrophobic matching.” In this configuration the insertion barrier drops below 8 kT, suggesting that such particles can spontaneously embed or translocate. Deviations from the matched length increase the barrier proportionally.
-
Parallel cylinders: Because the infinite‑cylinder model maximizes the contact area with the bilayer, a hydrophilic surface leads to a large unfavorable interaction. The calculated barrier exceeds 25 kT, effectively preventing insertion. Only when the cylinder surface is strongly hydrophobic does the barrier become comparable to the spherical case.
These results quantitatively validate the hydrophobic‑matching concept and demonstrate that particle shape, orientation, and surface patterning together dictate the thermodynamic feasibility of membrane insertion. The authors also show that the SCMF framework can compute the free‑energy profile ΔF*(p) as a function of particle position p, enabling estimation of kinetic barriers for insertion and extraction processes.
From a methodological perspective, the SCMF calculations for one‑dimensional (planar bilayer) and two‑dimensional (cylindrical symmetry) systems run on a 32‑core workstation in roughly one day, while full three‑dimensional simulations remain computationally demanding. The authors anticipate that further code optimization and parallelization will make 3D calculations tractable for larger, more realistic systems.
In conclusion, the paper provides a robust, computationally efficient tool for predicting how nanoscale objects interact with lipid membranes. By linking geometric parameters and surface chemistry to free‑energy landscapes, it offers practical guidance for designing drug‑delivery nanoparticles, biosensors, or antimicrobial agents with controlled membrane activity. Future extensions could incorporate patterned surfaces, membrane curvature, or dynamic lipid rearrangements to capture even more complex biological scenarios.
Comments & Academic Discussion
Loading comments...
Leave a Comment