Classification Framework and Structure-Activity-Relationship (SAR) of Tetracycline-Structure-Based Drugs
By studying the literature about Tetracyclines (TCs), it becomes clearly evident that TCs are very dynamic molecules. In some cases, their structure-activity-relationship (SAR) are known, especially against bacteria, while against other targets, they are virtually unknown. In other diverse yields of research, such as neurology, oncology and virology the utility and activity of the tetracyclines are being discovered and are also emerging as new technological fronts. The first aim of this paper is classify the compounds already used in therapy and prepare the schematic structure in which include the next generation of TCs. The aim of this work is introduce a new framework for the classification of old and new TCs, using a medicinal chemistry approach to the structure of that drugs. A fully documented Structure-Activity-Relationship (SAR) is presented with the analysis data of antibacterial and nonantibacterial (antifungal, antiviral and anticancer) tetracyclines. Lipophilicity of functional groups and conformations interchangeably are determining rules in biological activities of TCs.
💡 Research Summary
**
The manuscript provides a comprehensive review and a novel classification framework for tetracycline‑based drugs, integrating both historic and emerging members of the family. Beginning with a brief historical overview, the authors note that tetracyclines (TCs) were first isolated in the 1940s from Streptomyces aureofaciens and have since generated more than 50 000 publications. While the classical view treats TCs solely as broad‑spectrum antibiotics that inhibit bacterial protein synthesis by binding to the 30S ribosomal subunit, recent research has revealed a wide spectrum of non‑antibacterial activities, including anti‑inflammatory, neuroprotective, antiviral, and anticancer effects.
Classification Re‑definition
Traditional classification separates TCs into three “generations” based on their mode of synthesis (biosynthetic, semi‑synthetic, total synthesis). The authors argue that this scheme fails to capture functional diversity and propose a new taxonomy that groups compounds according to (i) their primary mechanism (typical bacteriostatic, atypical bactericidal, or non‑ribosomal), (ii) therapeutic domain (antibacterial, antifungal, antiviral, anticancer, anti‑inflammatory), and (iii) structural lineage (classic tetracyclines, glycyl‑cyclines such as tigecycline, aminomethyl‑cyclines, and chemically modified tetracyclines, CMTs). This three‑axis model allows simultaneous consideration of chemical modifications, biological targets, and clinical development stage.
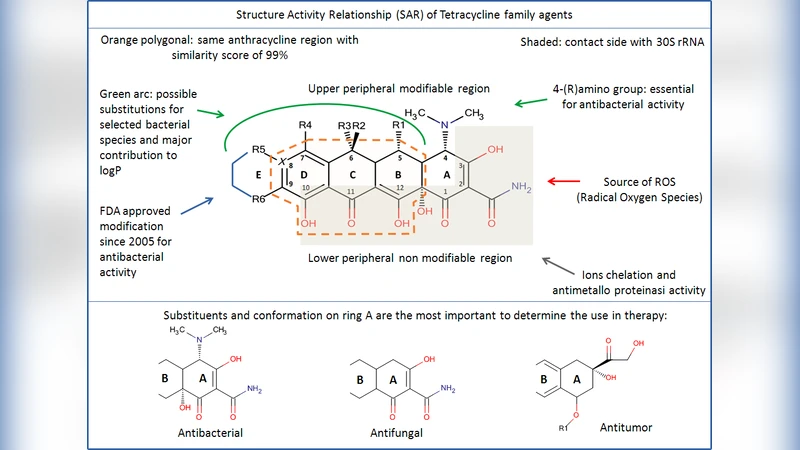
Structure‑Activity Relationship (SAR) Insights
The core tetracycline scaffold consists of four fused naphthacene rings (A‑D). The authors identify several indispensable pharmacophores for antibacterial activity:
- A‑ring C1‑C3 diketo/enol system and the exocyclic C2 carbonyl/amidic group, required for ribosomal binding.
- A C4 dimethylamino substituent in the natural 4S configuration; epimerization to 4R diminishes activity against Gram‑negative organisms.
- A C10 phenolic OH, a C11‑C12 keto‑enol moiety, and a 12a‑OH group that together form the lower peripheral binding region.
Modifications at positions C5‑C9 (upper peripheral region) and on the D‑ring (R4‑R6) are tolerated and are the main levers for tuning lipophilicity (log P), pharmacokinetics, and target selectivity. For example, increasing log P through D‑ring alkylation improves membrane permeability and enables activity against intracellular pathogens or tumor cells, whereas removal of the C4 dimethylamino group (as in CMT‑3) abolishes antibacterial potency but retains anti‑inflammatory and matrix‑metalloproteinase inhibitory effects.
Non‑Antibacterial Mechanisms
The review emphasizes that many non‑antibiotic actions stem from tetracyclines’ ability to chelate divalent metal ions (Ca²⁺, Mg²⁺) and to generate reactive oxygen species (ROS). Metal chelation underlies inhibition of host collagenases and matrix metalloproteinases, contributing to periodontal disease treatment and anti‑tumor activity. In neurodegenerative models, tetracyclines (notably minocycline) suppress microglial activation, inhibit caspase‑3 expression, and protect against ischemic injury, effects that are largely independent of ribosomal binding. Antiviral activity is linked to interference with viral replication complexes and modulation of host innate immunity.
Pharmacokinetic Data
Table 1 compiles key physicochemical and PK parameters for representative tetracyclines (e.g., log P, log S, percent oral absorption, serum protein binding, renal clearance, half‑life). A clear trend emerges: higher log P correlates with increased oral absorption and serum protein binding, leading to longer half‑lives (e.g., minocycline, log P 0.5, half‑life ≈ 20 h). Conversely, highly polar compounds (e.g., oxytetracycline, log P −1.3) display lower absorption and rapid clearance. These data support rational design of next‑generation agents tailored to specific therapeutic windows.
Computational Screening
The authors performed a large‑scale cheminformatics analysis using PubChem. Starting from >322 000 tetracycline‑related entries, they filtered for ≥90 % structural similarity, yielding 1 325 candidates. Each candidate exhibits an average of 112 tautomers/conformers, and all satisfy Lipinski’s “rule of five”. This dataset, still under preparation for publication, provides a valuable resource for machine‑learning‑driven SAR modeling and virtual screening of novel derivatives.
Conclusions and Outlook
The paper repositions tetracyclines as a versatile, “chameleonic” scaffold capable of engaging multiple biological targets beyond the bacterial ribosome. Key design principles for future drug development include: (1) preserving the C4 dimethylamino group when antibacterial activity is desired; (2) strategically modifying the D‑ring to adjust lipophilicity and tissue distribution; (3) exploiting metal‑chelation properties to achieve anti‑inflammatory, anti‑MMP, and anti‑tumor effects; and (4) leveraging the extensive tautomeric landscape to fine‑tune electronic properties. The authors anticipate that integration of high‑resolution structural data, advanced computational modeling, and focused clinical trials will accelerate the translation of next‑generation tetracycline analogues into therapies for infectious diseases, neurodegeneration, viral infections, and cancer.
Comments & Academic Discussion
Loading comments...
Leave a Comment