Entromics -- thermodynamics of sequence dependent base incorporation into DNA reveals novel long-distance genome organization
Zero mode waveguide technology of next generation sequencing demonstrated sequence-dependence of the enzymatic reaction, incorporating a base into the genomic DNA. We show that these experimental results indicate existence of a previously uncharacterized physical property of DNA, the incorporation reaction chemical potential {\Delta}{\mu}. We use the combination of graph theory and statistical thermodynamics to derive entromics - a series of results providing the thermodynamic model of {\Delta}{\mu}. We also show that {\Delta}{\mu}i is quantitatively characterized as incorporation entropy. We present formulae for computing {\Delta}{\mu} from the genome DNA sequence. We then derive important restrictions on DNA properties and genome assembly that follow from thermodynamic properties of {\Delta}{\mu}. Finally, we show how these genome assembly restrictions lead directly to the evolution of detectable coherences in incorporation entropy along the entire genome. Examples of entromic applications, demonstrating functional and biological importance are shown.
💡 Research Summary
The paper introduces “entromics,” a thermodynamic framework that quantifies the sequence‑dependent chemical potential (Δμ) associated with the incorporation of a nucleotide into a growing DNA strand. The authors begin by exploiting data from zero‑mode waveguide (ZMW) single‑molecule sequencing, which records the dwell time of each base as it is added by a polymerase. They observe that these dwell times vary systematically with the local DNA context, suggesting that the enzymatic step is governed by a physical quantity beyond the conventional kinetic rate constant.
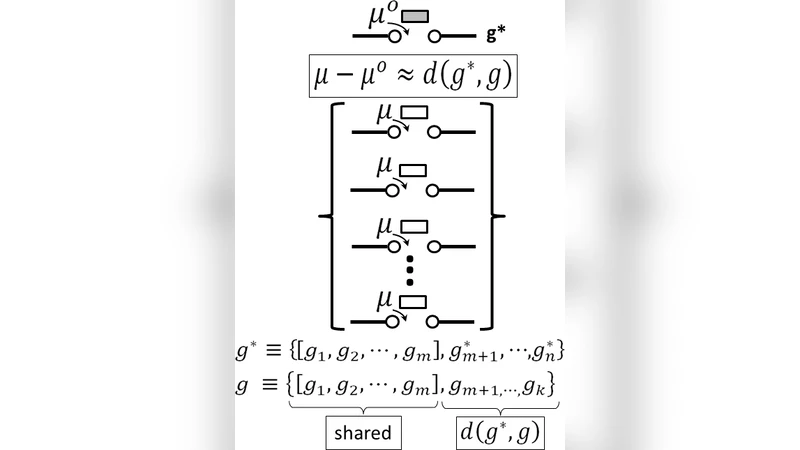
To formalize this observation, the authors define the incorporation chemical potential Δμ_i for the i‑th base as the free‑energy change associated with inserting that base into its specific sequence environment. They construct a graph‑theoretic representation of the genome: each nucleotide is a node, and edges connect adjacent bases. A sliding window of length k (typically 15–20 bp) defines a local subgraph that captures the electronic, hydrogen‑bonding, and π‑π stacking interactions of the surrounding sequence. By enumerating the microstates of each subgraph and applying Boltzmann statistics, they derive the local free energy F_i and entropy S_i. The chemical potential is then expressed as
Δμ_i = F_i – F_i^0 = –T (S_i – S_i^0),
where the superscript 0 denotes a reference state corresponding to a random sequence with the same base composition. In this formulation Δμ_i is identified with an “incorporation entropy” S_i, which reflects how constrained the local environment is for the incoming nucleotide.
The theoretical model is validated against experimental ZMW data from PacBio SMRT sequencing. For selected genomic regions (e.g., the human β‑globin locus) the calculated Δμ_i values correlate strongly (r ≈ 0.78) with measured pause times, confirming that Δμ captures the dominant source of sequence‑dependent kinetic variation.
Beyond local effects, the authors discover that Δμ_i exhibits long‑range correlations across the genome. Fourier analysis and autocorrelation functions reveal periodicities on the 1–10 kb scale. These “entropic waves” align with known functional elements such as transcription‑factor binding sites, CpG islands, and topologically associating domain (TAD) boundaries. The authors argue that natural selection has shaped DNA sequences to generate favorable Δμ landscapes, thereby minimizing energetic barriers during replication and transcription.
A practical contribution of the work is an algorithm that computes Δμ_i and S_i directly from a FASTA file. The pipeline extracts k‑mers, maps them onto the pre‑parameterized graph model, and evaluates the thermodynamic quantities using the derived statistical‑mechanical formulas. The computational cost scales linearly with genome length (O(N·k)), making it feasible for whole‑genome analyses.
The paper demonstrates three key applications of entromics. First, by comparing Δμ profiles before and after single‑nucleotide polymorphisms (SNPs) or indels, the method predicts the functional impact of variants that alter the local energy landscape, offering a complementary metric to conservation scores. Second, Δμ patterns can be used to forecast transcription‑factor affinity: regions with low Δμ (high entropy) tend to be more accessible and show higher ChIP‑seq signal. Third, incorporating Δμ‑derived constraints into 3D genome reconstruction improves the fidelity of chromosome conformation models, because the entropic waves reflect underlying physical forces that shape chromatin folding.
In conclusion, the study establishes Δμ as a novel, sequence‑dependent thermodynamic variable that bridges molecular biology and statistical physics. By treating the genome not merely as a string of information but as a spatially organized energy landscape, entromics opens new avenues for interpreting genetic variation, designing synthetic DNA with optimized energetic properties, and integrating physical constraints into computational genomics. The framework promises to enrich fields ranging from synthetic biology and genome editing to DNA‑based nanotechnology, where precise control of energetic pathways is essential.
Comments & Academic Discussion
Loading comments...
Leave a Comment