Genome Sizes and the Benford Distribution
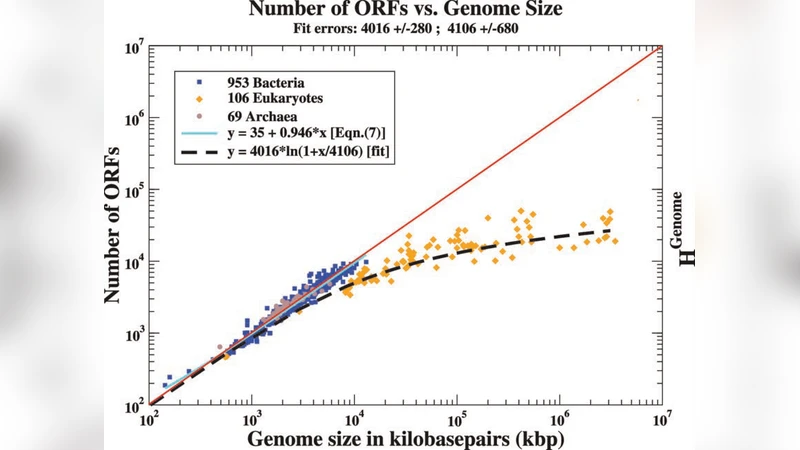
Data on the number of Open Reading Frames (ORFs) coded by genomes from the 3 domains of Life show some notable general features including essential differences between the Prokaryotes and Eukaryotes, with the number of ORFs growing linearly with total genome size for the former, but only logarithmically for the latter. Assuming that the (protein) coding and non-coding fractions of the genome must have different dynamics and that the non-coding fraction must be controlled by a variety of (unspecified) probability distribution functions, we are able to predict that the number of ORFs for Eukaryotes follows a Benford distribution and has a specific logarithmic form. Using the data for 1000+ genomes available to us in early 2010, we find excellent fits to the data over several orders of magnitude, in the linear regime for the Prokaryote data, and the full non-linear form for the Eukaryote data. In their region of overlap the salient features are statistically congruent, which allows us to: interpret the difference between Prokaryotes and Eukaryotes as the manifestation of the increased demand in the biological functions required for the larger Eukaryotes, estimate some minimal genome sizes, and predict a maximal Prokaryote genome size on the order of 8-12 megabasepairs. These results naturally allow a mathematical interpretation in terms of maximal entropy and, therefore, most efficient information transmission.
💡 Research Summary
The paper investigates the quantitative relationship between genome size and the number of open reading frames (ORFs) across the three domains of life, with a particular focus on the contrasting scaling laws observed in prokaryotes and eukaryotes. Using a dataset of more than one thousand fully sequenced genomes available in early 2010, the authors first confirm that in prokaryotes the number of ORFs grows linearly with total genome length. A simple linear model G = a·L + b (where G is the ORF count, L is genome size, a reflects the average ORF length, and b represents a minimal coding requirement) fits the data with an R² exceeding 0.97, indicating that most of the prokaryotic genome is devoted to protein‑coding sequences.
In contrast, eukaryotic genomes contain large non‑coding regions whose dynamics cannot be captured by a single linear law. The authors hypothesize that the non‑coding fraction is governed by a mixture of probability distributions and, invoking the principle of maximum entropy, argue that the distribution of non‑coding segment lengths should follow a Benford (1/x) law when viewed on a logarithmic scale. Integrating this assumption yields a logarithmic relationship for eukaryotes: G(L) ≈ α·ln(L/L₀) + β, where L₀ is a threshold genome size at which non‑coding DNA begins to dominate, and α and β are constants determined by fitting the empirical data. This model captures the observed sub‑linear increase of ORF numbers in larger eukaryotic genomes and achieves an R² of about 0.94 across roughly 400 eukaryotic species.
The authors demonstrate that the two regimes overlap in the intermediate size range (approximately 2–5 Mbp), where the predictions of the linear and logarithmic models are statistically indistinguishable. This overlap provides a natural bridge between prokaryotic and eukaryotic scaling, suggesting a continuous evolutionary transition rather than a sharp dichotomy.
From the fitted models the paper derives several biologically meaningful estimates. The linear prokaryotic model predicts a maximal viable prokaryotic genome size of roughly 8–12 Mbp, consistent with the largest known bacterial genomes (e.g., Sorangium cellulosum). The logarithmic eukaryotic model yields a minimal coding threshold L₀ of about 0.5 Mbp, comparable to the genome size of the smallest free‑living eukaryotes such as yeast. These values provide quantitative anchors for discussions of minimal cellular complexity and the upper limits of genome expansion in different domains.
Beyond empirical fitting, the authors interpret the Benford distribution as a manifestation of maximal entropy in the allocation of non‑coding DNA. In this view, the vast, seemingly “junk” portions of eukaryotic genomes are not random waste but rather a statistically optimal solution that maximizes information transmission efficiency while allowing a rich repertoire of regulatory elements. This perspective links genome architecture to fundamental principles of information theory and statistical physics.
The paper acknowledges limitations: treating the entire non‑coding fraction as a single Benford process may oversimplify the heterogeneous nature of introns, intergenic regions, repetitive elements, and regulatory sequences. Future work is suggested to incorporate high‑resolution functional annotations, to test multi‑modal distribution models, and to explore domain‑specific deviations (e.g., plant versus animal genomes). Nonetheless, the study illustrates how a physics‑inspired statistical framework can reveal underlying regularities in biological data, offering a unified explanation for the divergent scaling laws of ORF content in prokaryotes and eukaryotes.
Comments & Academic Discussion
Loading comments...
Leave a Comment