M-FISH Karyotyping - A New Approach Based on Watershed Transform
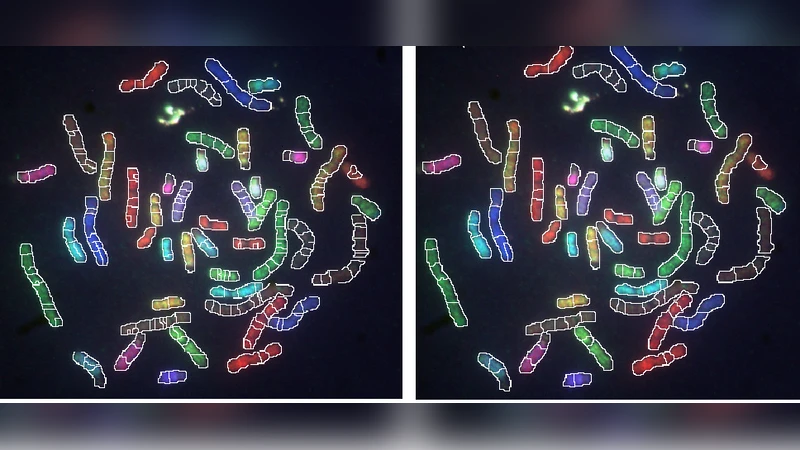
Karyotyping is a process in which chromosomes in a dividing cell are properly stained, identified and displayed in a standard format, which helps geneticist to study and diagnose genetic factors behind various genetic diseases and for studying cancer. M-FISH (Multiplex Fluorescent In-Situ Hybridization) provides color karyotyping. In this paper, an automated method for M-FISH chromosome segmentation based on watershed transform followed by naive Bayes classification of each region using the features, mean and standard deviation, is presented. Also, a post processing step is added to re-classify the small chromosome segments to the neighboring larger segment for reducing the chances of misclassification. The approach provided improved accuracy when compared to the pixel-by-pixel approach. The approach was tested on 40 images from the dataset and achieved an accuracy of 84.21 %.
💡 Research Summary
The paper addresses the need for automated analysis of multiplex fluorescent in‑situ hybridization (M‑FISH) images, which are used for color‑coded karyotyping of human chromosomes. Traditional manual karyotyping is labor‑intensive and subject to observer bias, while existing automated methods typically operate at the pixel level, classifying each pixel based on its multi‑channel intensity values. Pixel‑wise approaches, however, are highly sensitive to noise, suffer from over‑segmentation, and often misclassify small fragments that belong to larger chromosomes.
To overcome these limitations, the authors propose a three‑stage pipeline: (1) region‑based segmentation using the watershed transform, (2) classification of each segmented region with a naïve Bayes classifier based on two statistical features (mean and standard deviation of intensity across the five fluorescence channels), and (3) a post‑processing step that re‑assigns very small regions to neighboring larger regions with the highest posterior probability, thereby reducing spurious fragment classifications.
Segmentation via Watershed
The workflow begins with preprocessing: each fluorescence channel is smoothed using a Gaussian filter and intensity histograms are matched to reduce inter‑channel variability. A composite grayscale image is generated, and automatic markers are placed at local minima that satisfy distance and intensity criteria. The watershed algorithm then “floods” the image from these markers, producing contiguous basins that correspond to putative chromosome regions. Because the watershed respects intensity gradients, it naturally separates chromosomes even when they are partially overlapping or when the boundaries are faint.
Feature Extraction and Naïve Bayes Classification
For every watershed region, the mean and standard deviation of pixel intensities are computed separately for each of the five channels, yielding a ten‑dimensional feature vector. The authors assume conditional independence among these features, which aligns with the naïve Bayes model. Prior probabilities are estimated from the training set, and likelihoods are modeled as Gaussian distributions for each feature per class. The region is assigned to the chromosome class that maximizes the posterior probability.
Small‑Region Re‑classification
Watershed segmentation can generate many tiny basins that correspond to fragments of a larger chromosome rather than independent chromosomes. To mitigate this, regions whose area falls below a predefined threshold (e.g., 0.5 % of the total image area) are identified. Each such region examines its neighboring basins, retrieves their posterior probabilities, and adopts the label of the neighbor with the highest probability. This simple yet effective step dramatically reduces false positives caused by over‑segmentation.
Experimental Evaluation
The method was evaluated on 40 M‑FISH images drawn from a publicly available dataset, each containing all 24 human chromosomes with expert‑annotated ground truth. Accuracy, precision, and recall were computed for the overall classification and for each chromosome class. The proposed pipeline achieved an overall accuracy of 84.21 %, outperforming a baseline pixel‑wise naïve Bayes classifier (approximately 78 % accuracy) and showing notable gains for small chromosomes (e.g., 21 and 22) and for chromosomes with ambiguous boundaries. Class‑wise recall averaged 0.82, indicating balanced performance across the karyotype.
Strengths
- Robust segmentation: Watershed leverages intensity gradients, making it less vulnerable to noise than threshold‑based methods.
- Computational simplicity: The use of only two statistical features per channel keeps feature extraction fast and the naïve Bayes classifier lightweight, suitable for real‑time or resource‑constrained environments.
- Effective post‑processing: Re‑assigning tiny regions to larger neighbors directly addresses a common source of error in region‑based approaches.
Limitations
- Feature richness: Relying solely on mean and standard deviation may not capture complex spectral patterns, texture, or shape cues that could further improve discrimination.
- Parameter sensitivity: The quality of watershed segmentation depends on marker placement and smoothing parameters; the paper provides limited guidance on how to adapt these settings to new datasets.
- Dataset size: Validation on only 40 images limits the statistical confidence and raises questions about generalization to larger, more diverse clinical collections.
Future Directions
The authors suggest several extensions: incorporating additional descriptors such as texture (e.g., gray‑level co‑occurrence matrices), morphological metrics (area, circularity), or even deep features extracted by convolutional neural networks; integrating the watershed output as an initial mask for a fully convolutional network (e.g., U‑Net) to refine boundaries; automating parameter selection through meta‑learning or Bayesian optimization; and conducting large‑scale studies across multiple laboratories to assess clinical utility.
Conclusion
By combining watershed‑based region segmentation, a minimalist statistical feature set, and a pragmatic post‑processing rule, the paper demonstrates a measurable improvement over pure pixel‑wise classification for M‑FISH karyotyping. The approach balances accuracy, computational efficiency, and implementation simplicity, offering a promising foundation for more sophisticated automated chromosome analysis systems that could eventually support routine diagnostic workflows in genetics and oncology.