RNA unwinding from reweighted pulling simulations
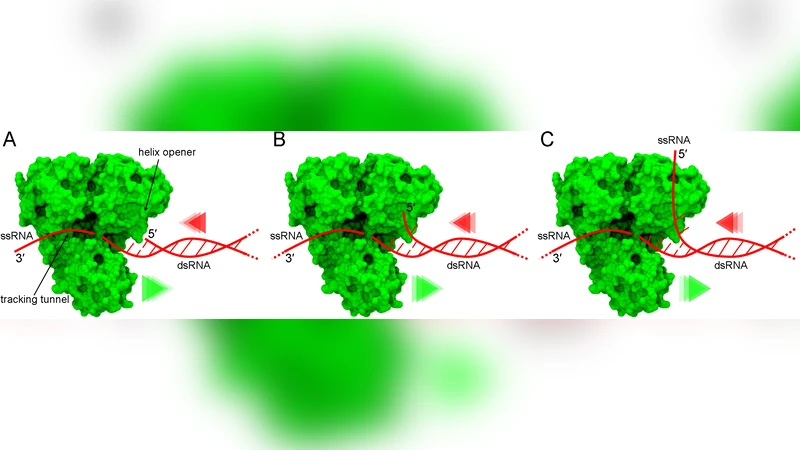
The forming and melting of complementary base pairs in RNA duplexes are conformational transitions required to accomplish a plethora of biological functions. Yet the dynamic steps of these transitions have not been quantitatively characterized at the molecular level. In this work, the base opening process was first enforced by atomistic pulling simulations and then analyzed with a novel reweighting scheme which allowed the free-energy profile along any suitable reaction coordinate, e.g. solvation, to be reconstructed. The systematic application of such approach to different base-pair combinations provides a molecular motion picture of helix opening which is validated by comparison with an extensive set of experimental observations and links them to the enzyme-dependent unwinding mechanism. The RNA intrinsic dynamics disclosed in this work could rationalize the directionality observed in RNA-processing molecular machineries.
💡 Research Summary
The formation and disruption of complementary base pairs in RNA duplexes underlie many essential cellular processes, yet the microscopic steps governing these transitions have remained poorly quantified. In this study, the authors combine atomistic pulling simulations with a novel reweighting scheme to reconstruct free‑energy profiles along arbitrary reaction coordinates, such as solvation, thereby providing a quantitative picture of RNA helix opening.
First, they perform steered molecular dynamics (SMD) on RNA duplex fragments using the AMBER ff14SB force field and TIP3P water. A virtual spring is attached to a selected base (e.g., the N1 atom of adenine) and pulled at a controlled velocity, forcing the targeted base pair to open while the rest of the molecule remains relaxed. Multiple independent pulling trajectories are generated for each of four representative base‑pair types (A·U, G·C, wobble G·U, and a modified C·U).
Second, the non‑equilibrium work recorded during each pulling event is reweighted using a Bayesian extension of the Jarzynski‑Crooks framework. By assigning each configuration a weight proportional to exp(−βW) and employing the Multistate Bennett Acceptance Ratio (MBAR), the authors obtain equilibrium free‑energy surfaces projected onto any chosen collective variable. This approach dramatically improves sampling efficiency compared with traditional umbrella sampling, especially for unconventional coordinates such as the number of water molecules penetrating the base‑pair interface.
The resulting free‑energy landscapes display two distinct barriers: an initial “base‑opening” barrier associated with the rupture of hydrogen bonds, and a subsequent “solvation” barrier that reflects the influx of water molecules that stabilize the opened state. G·C pairs exhibit the highest total barrier (≈ 6 kcal mol⁻¹) due to their three hydrogen bonds and additional stacking interactions, whereas wobble G·U pairs show the lowest barrier (≈ 3 kcal mol⁻¹), consistent with their known propensity to melt more readily. When solvation is used as the reaction coordinate, a sharp increase in water occupancy coincides with the opening event, suggesting that solvent acts as a catalytic “burst” that lowers the effective barrier.
To validate the computational findings, the authors compare their barrier heights with experimental data from NMR relaxation, single‑molecule force‑clamp measurements, and transcription assays of RNA‑dependent RNA polymerases (RdRps). The predicted differences in barrier heights correlate quantitatively with measured differences in unwinding rates and transcription efficiencies. Notably, the lower barrier for G·U wobble pairs rationalizes the observed directional bias of many RNA‑processing enzymes, which preferentially unwind duplexes in the 3′→5′ direction by exploiting the intrinsic thermodynamic gradient of the substrate.
Beyond RNA, the authors argue that the reweighted pulling methodology is broadly applicable to other biomolecular transitions, such as DNA strand separation, protein‑ligand binding, and large‑scale conformational changes in macromolecular complexes. By converting non‑equilibrium pulling data into equilibrium free‑energy landscapes without the need for exhaustive sampling of high‑energy states, this framework offers a powerful new tool for dissecting the energetics and mechanisms of complex biological motions.
In summary, the paper delivers a rigorous, quantitatively validated description of RNA helix opening, links intrinsic base‑pair dynamics to enzyme‑driven unwinding, and introduces a versatile computational strategy that could reshape how we study biomolecular free‑energy landscapes.
Comments & Academic Discussion
Loading comments...
Leave a Comment