Cancer gene prioritization by integrative analysis of mRNA expression and DNA copy number data: a comparative review
A variety of genome-wide profiling techniques are available to probe complementary aspects of genome structure and function. Integrative analysis of heterogeneous data sources can reveal higher-level interactions that cannot be detected based on individual observations. A standard integration task in cancer studies is to identify altered genomic regions that induce changes in the expression of the associated genes based on joint analysis of genome-wide gene expression and copy number profiling measurements. In this review, we provide a comparison among various modeling procedures for integrating genome-wide profiling data of gene copy number and transcriptional alterations and highlight common approaches to genomic data integration. A transparent benchmarking procedure is introduced to quantitatively compare the cancer gene prioritization performance of the alternative methods. The benchmarking algorithms and data sets are available at http://intcomp.r-forge.r-project.org
💡 Research Summary
The paper presents a systematic comparative review of twelve publicly available algorithms that integrate genome‑wide gene expression (GE) and DNA copy‑number (CN) data for the purpose of prioritizing cancer‑related genes. Recognizing that many cancer studies aim to identify genomic regions where CN alterations drive changes in transcription, the authors first surveyed the literature and the Bioconductor repository using keywords such as “gene expression”, “copy number”, and “integration”. This effort yielded twelve methods spanning several methodological families: two‑step approaches that first call CN alterations and then test differential expression, regression‑based models (both linear and non‑linear), correlation‑based techniques (including DR‑Correlate and modified Ortiz‑Estevez), canonical correlation analysis (CCA) and its regularized or sparse variants, and latent‑variable models such as pint/simcca, PREDA/SODEGI R, and related probabilistic CCA frameworks.
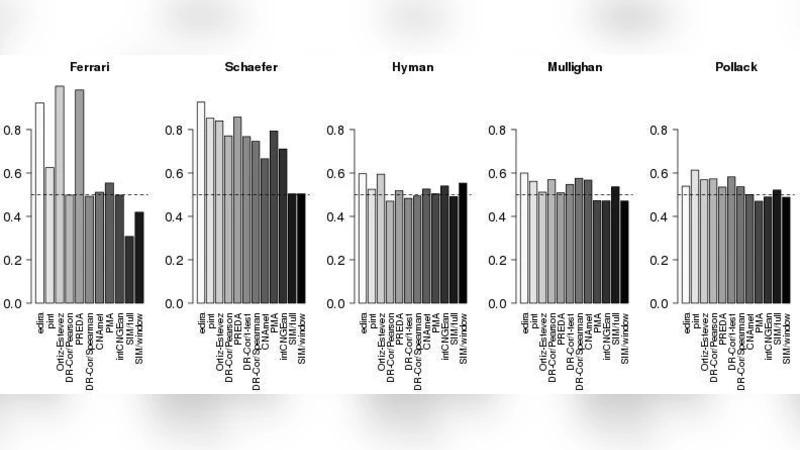
To evaluate these methods, the authors constructed a transparent benchmarking pipeline called intcomp, implemented in R and made publicly available. Performance was assessed on two simulated data sets—‘Schäfer’, which mimics block‑wise CN/GE alterations with varying noise levels, and ‘Ferrari’, derived from a renal‑cell carcinoma dataset with artificially introduced CN‑GE changes—as well as three real cancer case studies: two breast‑cancer microarray cohorts (Hyman and Pollack) and an acute lymphoblastic leukemia (ALL) cohort (Mullighan). For the real data, curated lists of known breast‑cancer genes and the Cancer Gene Census (leukemia genes) served as the gold‑standard reference.
Three complementary metrics were used: (i) the area under the ROC curve (AUC) for the full ranked gene list, (ii) the true‑positive rate among the top‑N genes (N = 200, 100, 50, 20), reflecting the practical need to select a manageable set of candidates for experimental validation, and (iii) computational runtime on a 64‑bit Linux server with 32 GB RAM. The authors applied each algorithm with default parameters, making minor adjustments where necessary (e.g., handling of missing p‑values, segmentation of CN data, or permutation settings).
The results reveal clear differences among the methods. In terms of median AUC across all five data sets, edira achieved the highest ranking, followed closely by Ortiz‑Estevez and pint/simcca. When focusing on the top‑200 genes, pint/simcca showed the best true‑positive rate, with edira, Ortiz‑Estevez, and PREDA/SODEGI R also performing strongly. Execution‑time analysis highlighted that edira and PMA were the fastest (under one minute for all datasets), while permutation‑heavy methods such as CNAmet, DR‑Correlate, intCNGEan, and PREDA required substantially longer runtimes (up to several hours for the most demanding settings). The authors note that some algorithms (e.g., those originally designed for region‑level detection) performed exceptionally well on the simulated data because the simulations were constructed to match their modeling assumptions, cautioning against over‑interpretation of simulation‑only results.
The discussion emphasizes that while simulation studies suggest high theoretical performance, real‑world data present greater challenges: the curated cancer‑gene lists are incomplete, noise levels are higher, and sample sizes vary. Consequently, the authors advocate for a nuanced selection of integration methods based on the specific characteristics of the dataset, the desired balance between sensitivity and specificity, and computational resources. The intcomp package provides a reproducible framework for future benchmarking, allowing researchers to add new algorithms, datasets, or evaluation criteria with minimal effort.
In summary, this review fills a gap left by previous surveys by delivering a quantitative, reproducible comparison of CN‑GE integration tools. It demonstrates that methods such as edira, Ortiz‑Estevez, pint/simcca, and PREDA/SODEGI R generally outperform others in cancer gene prioritization, but the optimal choice remains context‑dependent. The publicly released benchmarking pipeline and dataset collection constitute valuable resources for the community, facilitating more informed methodological decisions and accelerating the discovery of functional cancer drivers.
Comments & Academic Discussion
Loading comments...
Leave a Comment