CytoITMprobe: a network information flow plugin for Cytoscape
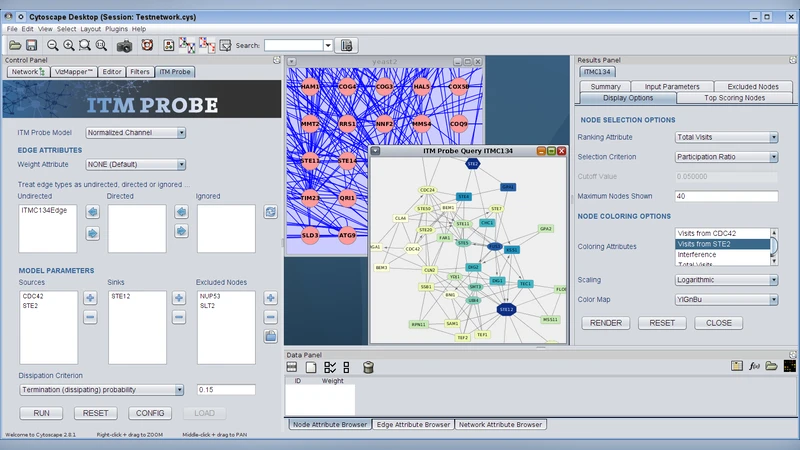
To provide the Cytoscape users the possibility of integrating ITM Probe into their workflows, we developed CytoITMprobe, a new Cytoscape plugin. CytoITMprobe maintains all the desirable features of ITM Probe and adds additional flexibility not achievable through its web service version. It provides access to ITM Probe either through a web server or locally. The input, consisting of a Cytoscape network, together with the desired origins and/or destinations of information and a dissipation coefficient, is specified through a query form. The results are shown as a subnetwork of significant nodes and several summary tables. Users can control the composition and appearance of the subnetwork and interchange their ITM Probe results with other software tools through tab-delimited files. The main strength of CytoITMprobe is its flexibility. It allows the user to specify as input any Cytoscape network, rather than being restricted to the pre-compiled protein-protein interaction networks available through the ITM Probe web service. Users may supply their own edge weights and directionalities. Consequently, as opposed to ITM Probe web service, CytoITMprobe can be applied to many other domains of network-based research beyond protein-networks. It also enables seamless integration of ITM Probe results with other Cytoscape plugins having complementary functionality for data analysis.
💡 Research Summary
CytoITMprobe is a Cytoscape plug‑in that brings the ITM Probe information‑flow framework into the Cytoscape environment, thereby overcoming the limitations of the original web service which only supports a few pre‑compiled protein‑protein interaction (PPI) networks. The plug‑in accepts any weighted, directed graph that the user can construct or import into Cytoscape, together with a set of boundary nodes (origins and/or destinations) and a dissipation (damping) coefficient µ (0 < µ < 1). Users specify these parameters through a graphical query form; the plug‑in then forwards the request either to a locally installed Python implementation of ITM Probe or to a remote web service.
ITM Probe implements three mathematically equivalent models based on discrete‑time random walks (or diffusion) on directed graphs:
-
Absorbing model – boundary nodes are sinks. For each transient node i and each sink k, the algorithm computes Fₖᵢ, the expected number of visits to sink k by walkers that start at i (or equivalently the probability that a walk from i terminates at k). In the absence of damping the probabilities sum to one; with damping they sum to less than one, reflecting loss of walkers.
-
Emitting model – boundary nodes are sources. For each source s and each transient node i, the algorithm returns Hₛᵢ, the expected number of visits to i by walkers that originate at s. Walkers may either dissipate or return to the source, which forms an emitting boundary.
-
Channel model – combines sources and sinks. For each source s, transient node i and sink k it computes Φₛᵢₖ = Hₛᵢ · Fₖᵢ, the average number of visits to i by walkers that start at s and terminate at k. The plug‑in reports a normalized quantity ˆΦₛᵢᴷ that removes the effect of damping on the denominator, thus highlighting the most likely paths from sources to sinks.
Mathematically, the weighted adjacency matrix W and damping factor µ define a transition matrix P where Pᵢⱼ = µ·Wᵢⱼ / ∑ⱼWᵢⱼ. Restricting P to transient nodes yields P_TT; the fundamental matrix G = (I − P_TT)⁻¹ is then used to compute F and H via simple matrix products. These calculations reduce to solving large, sparse, non‑symmetric linear systems. The Python back‑end relies on NumPy and SciPy; for direct sparse solves it uses SuperLU by default and optionally UMFPACK, which can be faster when linked against optimized BLAS libraries.
The user interface, written in Java using the Cytoscape API, consists of a query form, a results viewer, and an “ITM subnetwork” panel. After a query is executed, the plug‑in stores all results as node and edge attributes of the original Cytoscape network. Consequently, users can immediately apply Cytoscape’s layout algorithms, visual styles, or other plug‑ins (e.g., functional enrichment, clustering) to the ITM subnetwork. Results can also be exported as tab‑delimited files for downstream analysis.
Key advantages of CytoITMprobe are:
- Flexibility of input – any directed, weighted network can be analyzed, not just the curated PPI datasets of the web service.
- Control over locality – the damping coefficient µ determines how far information can travel; low µ emphasizes local neighborhoods, while higher µ allows more global exploration. In the channel model, µ also controls the “width” of the channel between sources and sinks.
- Seamless Cytoscape integration – results become native Cytoscape objects, enabling rich visualizations, easy combination with other analyses, and reproducible workflows.
- Dual execution modes – users may run the heavy linear‑algebra back‑end locally (for speed on large graphs) or rely on the remote web service (no installation required).
The paper demonstrates the three models on a small example network (19 nodes, 44 edges) and illustrates how damping shapes the visitation patterns. The emitting model highlights nodes frequently visited from a source, the absorbing model highlights nodes that efficiently funnel walkers to sinks, and the channel model reveals the most probable directed paths between designated origins and destinations.
In conclusion, CytoITMprobe extends the powerful random‑walk‑based information‑flow analysis of ITM Probe to the versatile Cytoscape platform, making it applicable not only to protein interaction studies but also to any domain where directed weighted networks are of interest (e.g., signaling pathways, metabolic networks, social or communication networks). By providing a mathematically rigorous yet user‑friendly tool, it enables researchers to explore context‑specific network neighborhoods, adjust the scale of information propagation, and integrate the results with the broad ecosystem of Cytoscape plug‑ins for downstream biological interpretation.
Comments & Academic Discussion
Loading comments...
Leave a Comment