Universal geometrical factor of protein conformations as a consequence of energy minimization
The biological activity and functional specificity of proteins depend on their native three-dimensional structures determined by inter- and intra-molecular interactions. In this paper, we investigate the geometrical factor of protein conformation as a consequence of energy minimization in protein folding. Folding simulations of 10 polypeptides with chain length ranging from 183 to 548 residues manifest that the dimensionless ratio (V/(A
💡 Research Summary
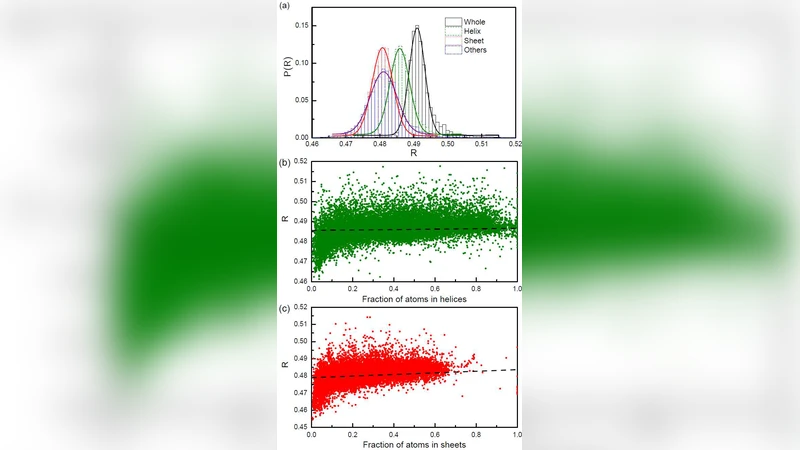
The paper investigates a universal geometric property of protein structures that emerges as a consequence of energy minimization during folding. Using the SMMP force field, the authors assign van der Waals radii to each atom (C 1.70 Å, N 1.55 Å, O 1.52 Å, H 1.20 Å, etc.) and compute the total van der Waals volume (V) and surface area (A) of a protein model. They then define a dimensionless ratio R = V/(A·
First, ten polypeptides with lengths ranging from 183 to 548 residues are subjected to steepest‑descent followed by conjugate‑gradient energy minimization. Starting from random coil conformations, the ratio R drops from ~0.35–0.40 and rapidly converges to ~0.49 within ~10⁴ minimization steps, after which it remains stable within ±0.005. The convergence occurs irrespective of chain length, secondary‑structure composition, or the proportion of hydrophobic versus hydrophilic residues, indicating that the minimization drives the system toward a common geometric optimum.
To test whether this behavior is present in real proteins, the authors analyze a large, non‑redundant set of >5,000 high‑resolution crystal structures from the Protein Data Bank. For each structure they compute V, A, and
The authors interpret the result in terms of a physical optimization problem: a body that minimizes surface energy for a given volume tends toward a spherical shape, which has the smallest possible A/V ratio. Proteins, despite their complex topology and heterogeneous interactions, appear to obey a similar principle when they reach a low‑energy native state. The ratio therefore reflects a balance between internal packing efficiency and exposure of surface residues, a balance that is largely independent of the detailed amino‑acid composition.
From a practical standpoint, the universal ratio can serve as a quick quality‑control metric for computational models, de‑novo designs, and homology‑based predictions. By checking whether a candidate structure yields R ≈ 0.49, modelers can assess whether the geometry is physically plausible before investing in more expensive simulations or experimental validation. The authors propose that incorporating this geometric constraint into protein‑design algorithms could improve the stability and functional reliability of engineered proteins.
In conclusion, the study establishes four key points: (1) energy minimization drives diverse polypeptide chains toward a common V/(A·