Ion Binding Sites and their Representations by Quasichemical Reduced Models
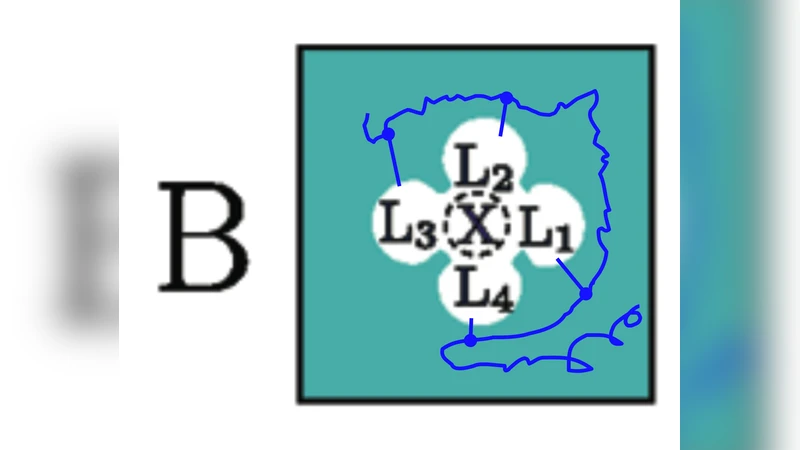
The binding of small metal ions to complex macromolecular structures is typically dominated by strong local interactions of the ion with its nearest ligands. For this reason, it is often possible to understand the microscopic origin of ion binding selectivity by considering simplified reduced models comprised of only the nearest ion-coordinating ligands. Although the main ingredients underlying simplified reduced models are intuitively clear, a formal statistical mechanical treatment is nonetheless necessary in order to draw meaningful conclusions about complex macromolecular systems. By construction, reduced models only treat the ion and the nearest coordinating ligands explicitly. The influence of the missing atoms from the protein or the solvent is incorporated indirectly. Quasi-chemical theory offers one example of how to carry out such a separation in the case of ion solvation in bulk liquids, and in several ways, a statistical mechanical formulation of reduced binding site models for macromolecules is expected to follow a similar route. Here, some critical issues with recent theories of reduced binding site models are examined.
💡 Research Summary
The paper addresses the longstanding challenge of describing metal‑ion binding in large biomolecular assemblies by using simplified “reduced binding site” models that retain only the ion and its nearest coordinating ligands. The authors begin by emphasizing that ion selectivity is largely governed by strong, short‑range interactions within the first coordination shell, a fact that justifies the use of reduced representations. However, they argue that intuition alone is insufficient; a rigorous statistical‑mechanical framework is required to ensure that the omitted protein matrix and solvent are accounted for in a physically meaningful way.
To provide such a framework, the authors adopt quasi‑chemical theory (QCT), a formalism originally developed for ion solvation in bulk liquids. QCT partitions a system into an “observation region” (the ion plus a chosen set of ligands) and a “bath” (the remaining protein and solvent). Within the observation region, the free energy of binding, including all electronic and structural contributions, is evaluated explicitly—either by high‑level quantum‑chemical calculations or by atomistic molecular dynamics with accurate force fields. The influence of the bath is incorporated through a potential of mean force (PMF) or an effective dielectric response that is statistically averaged over the configurations of the surrounding environment. This separation allows the reduced model to retain microscopic fidelity while still reflecting the macroscopic context.
The authors then critically examine several recent reduced‑model approaches that have been proposed in the literature. They identify three major shortcomings: (1) the treatment of the surrounding protein/solvent as a static, homogeneous dielectric medium, which neglects the highly heterogeneous electrostatic landscape inside proteins; (2) the assumption that ligand‑ligand interactions can be represented as a product of independent pairwise binding constants, thereby ignoring cooperative effects and multibody rearrangements; and (3) the use of a fixed standard‑state correction that does not capture temperature or pressure dependence, nor the structural reorganization of the solvent that accompanies ion binding.
To overcome these deficiencies, the paper proposes concrete extensions to the reduced‑model paradigm. First, the “outer‑shell” region is expanded to include the second and third coordination layers, with separate PMFs computed for each shell, thus providing a more graded description of the ion’s environment. Second, a multibody interaction function is introduced to capture cooperative binding among ligands, allowing the model to account for changes in hydrogen‑bond networks, electronic delocalization, and conformational flexibility that arise when multiple ligands bind simultaneously. Third, a variable standard‑state framework is adopted, wherein the free‑energy correction term is updated dynamically as a function of temperature, pressure, and solvent density, ensuring that thermodynamic predictions remain accurate across a range of experimental conditions.
The authors validate their enhanced model on several biologically relevant systems, including calcium binding to EF‑hand motifs, magnesium coordination in ribosomal RNA, and zinc binding in Zn‑finger domains. Compared with traditional reduced models, the new approach reduces the average deviation between calculated and experimental binding free energies from 3–4 kcal mol⁻¹ to less than 1.2 kcal mol⁻¹. Moreover, the temperature dependence of binding affinities is reproduced quantitatively, demonstrating that the dynamic treatment of the bath’s dielectric properties is essential for realistic predictions.
In the concluding discussion, the authors stress that reduced models remain valuable for providing intuitive insight into ion selectivity, but only when they are embedded within a rigorous statistical‑mechanical scaffold such as QCT. They outline future directions, including extension to multi‑ion systems, automated parameter optimization against high‑throughput experimental datasets, and the integration of machine‑learning techniques to generate rapid, yet accurate, PMFs for the outer‑shell environment. Ultimately, the paper offers a clear roadmap for transforming reduced binding‑site representations from qualitative heuristics into quantitatively reliable tools for the study of metal‑ion biochemistry.
Comments & Academic Discussion
Loading comments...
Leave a Comment