Effects of confinement on thermal stability and folding kinetics in a simple Ising-like model
In cellular environment, confinement and macromulecular crowding play an important role on thermal stability and folding kinetics of a protein. We have resorted to a generalized version of the Wako-Saito-Munoz-Eaton model for protein folding to study the behavior of six different protein structures confined between two walls. Changing the distance 2R between the walls, we found, in accordance with previous studies, two confinement regimes: starting from large R and decreasing R, confinement first enhances the stability of the folded state as long as this is compact and until a given value of R; then a further decrease of R leads to a decrease of folding temperature and folding rate. We found that in the low confinement regime both unfolding temperatures and logarithm of folding rates scale as R-{\gamma} where {\gamma} values lie in between 1.42 and 2.35.
💡 Research Summary
In this work the authors investigate how spatial confinement influences the thermal stability and folding kinetics of proteins by employing a generalized version of the Wako‑Saito‑Muñoz‑Eaton (WSME) model, an Ising‑like statistical‑mechanical description of protein folding. Six systems are examined: three idealized structures (a ten‑residue α‑helix, a two‑strand β‑sheet and a three‑strand β‑sheet) and three real proteins (a three‑helix bundle (PDB 1PRB), protein G (PDB 2GB1) and the C‑terminal β‑hairpin of protein G). The proteins are confined between two perfectly repulsive parallel walls separated by a distance 2R, and the effect of varying R is studied.
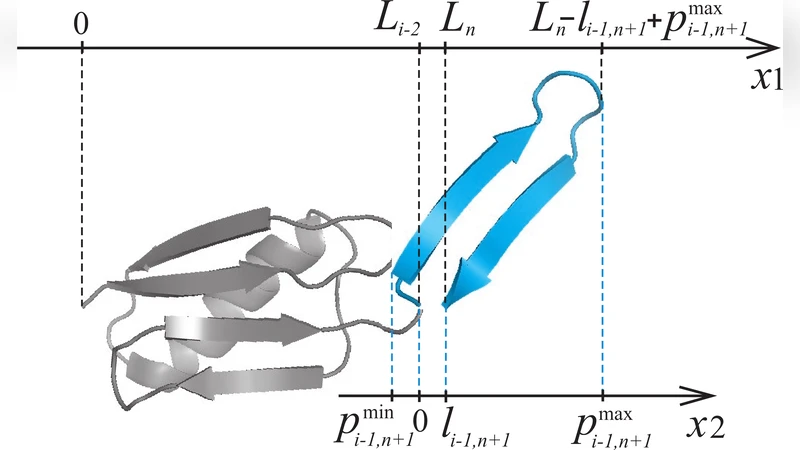
The WSME model represents each residue by a binary variable m_k (1 = native, 0 = unfolded). Native contacts contribute a negative energy –ε_ij only when all residues between i and j are also native. The Hamiltonian includes these contact terms and, when a force is applied, a term proportional to the end‑to‑end length L. To incorporate confinement, the authors introduce the concept of an “effective length”, i.e., the distance between the two farthest residues in a given configuration. For each native stretch they compute the maximal and minimal possible extensions (p_max and p_min) and embed Heaviside step functions into the recursive calculation of the constrained partition function. Translational freedom is accounted for by moving the cage relative to the N‑terminus and summing over all admissible positions, thereby correctly counting configurations that differ only by overall translation.
Thermodynamic quantities (Helmholtz free energy, specific heat, average fraction of native residues) are computed for a wide range of cage sizes, from just above the minimal unfolded length (~4 Å) up to twice the fully extended length. Two confinement regimes emerge. In the “weak confinement” regime (2R > L_N^eff, where L_N^eff is the effective length of the native state), the cage excludes the most expanded conformations of the unfolded basin while still accommodating the folded structure. This reduces conformational entropy, stabilizing the native basin relative to the unfolded basin. The effect is structure‑dependent: the three‑helix bundle shows modest stabilization of both native and unfolded basins, with a slightly larger gain for the native state; the β‑hairpin of protein G experiences destabilization of both basins, the unfolded one more strongly; the ideal α‑helix, whose unfolded effective length exceeds its native effective length, shows essentially no stabilization. In the “strong confinement” regime (2R < L_N^eff) the cage becomes too small even for the native conformation, leading to a sharp decrease in the folding temperature.
Kinetic analysis is performed via Monte‑Carlo simulations that measure the folding rate k_f as a function of R. Both the shift in folding temperature ΔT_f and the logarithm of the folding rate ln k_f follow a power‑law dependence on the cage size, ΔT_f ∝ R^–γ and ln k_f ∝ R^–γ. The exponent γ varies between 1.42 and 2.35 across the six systems. These values lie between the theoretical predictions for an ideal Gaussian chain (γ = 2) and for an excluded‑volume chain (γ ≈ 5/3 in one dimension, 15/4 in three dimensions), and are consistent with previous simulation studies that reported γ ≈ 3.25 for a cylindrical cage. The spread of γ reflects the interplay of native compactness, contact network topology, and the translational entropy contribution.
The paper’s main contributions are: (1) a rigorous extension of the exactly solvable WSME model to include hard‑wall confinement, preserving analytical tractability; (2) a quantitative demonstration that confinement can both enhance and suppress protein stability and folding rates, depending on the relative size of the cage and the native structure; (3) the identification of a scaling regime for folding thermodynamics and kinetics with exponents that bridge existing theoretical and experimental results. These findings deepen our understanding of how cellular crowding and confinement—such as in ribosomal exit tunnels, chaperonin cavities, or synthetic nanopores—modulate protein folding, and they provide a useful framework for interpreting experiments that probe folding under nanoconfinement.
Comments & Academic Discussion
Loading comments...
Leave a Comment