Parameter Estimation in multiple-hidden i.i.d. models from biological multiple alignment
In this work we deal with parameter estimation in a latent variable model, namely the multiple-hidden i.i.d. model, which is derived from multiple alignment algorithms. We first provide a rigorous formalism for the homology structure of k sequences related by a star-shaped phylogenetic tree in the context of multiple alignment based on indel evolution models. We discuss possible definitions of likelihoods and compare them to the criterion used in multiple alignment algorithms. Existence of two different Information divergence rates is established and a divergence property is shown under additional assumptions. This would yield consistency for the parameter in parametrization schemes for which the divergence property holds. We finally extend the definition of the multiple-hidden i.i.d. model and the results obtained to the case in which the sequences are related by an arbitrary phylogenetic tree. Simulations illustrate different cases which are not covered by our results.
💡 Research Summary
The paper addresses the statistical problem of estimating parameters in a latent‑variable framework that arises naturally from biological multiple‑sequence alignment (MSA) algorithms. The authors introduce the “multiple‑hidden i.i.d. model,” a probabilistic representation of the indel (insertion–deletion) process that underlies most progressive alignment methods.
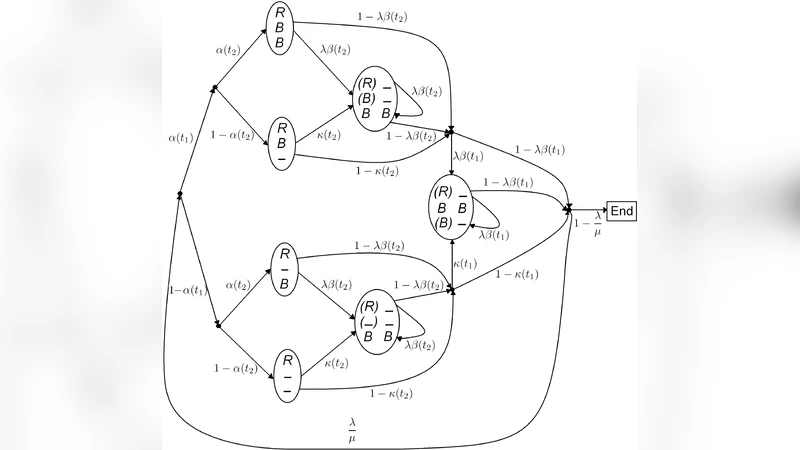
First, they formalize the homology structure for k sequences that share a common ancestor arranged in a star‑shaped phylogenetic tree. Each descendant sequence is generated by an independent i.i.d. block process that inserts and deletes characters along the lineage. This construction yields a hidden‑variable model: the observable aligned columns are produced from an unobserved indel path.
Second, the paper distinguishes two likelihood concepts. The complete‑likelihood incorporates the full hidden indel history, while the partial‑likelihood (the criterion actually optimized in many MSA programs) integrates out the hidden variables and depends only on the observed alignment. The authors prove that these two likelihoods are not equivalent for parameter estimation; in particular, the partial‑likelihood may fail to separate distinct parameter values.
The core theoretical contribution is the establishment of two information divergence rates. Under mild regularity conditions—most notably exponential tails for indel length distributions and bounded insertion probabilities—the authors show that both divergence rates are strictly positive for any parameter vector that differs from the true generating parameters. This “divergence property” implies that the Kullback–Leibler divergence between the true model and any misspecified model grows linearly with sequence length, guaranteeing consistency of the maximum likelihood estimator (MLE) when the appropriate likelihood is used.
Third, the authors extend the model from the star topology to an arbitrary phylogenetic tree. Each internal node is assigned its own independent indel process, and the homology structure is defined recursively along the tree. The same two divergence rates can be derived, but additional assumptions (e.g., uniformity of indel mechanisms across branches) become necessary to preserve the divergence property.
To assess practical relevance, extensive simulations are performed. Scenarios with high insertion rates or long insertion lengths violate the regularity assumptions, leading to a breakdown of the divergence property and biased MLEs. Conversely, under realistic evolutionary regimes the assumptions hold, and the estimated parameters converge to the true values. The simulations also compare the performance of estimators based on complete versus partial likelihoods, confirming the theoretical prediction that the complete‑likelihood yields consistent estimates while the partial‑likelihood may be inconsistent in certain regimes.
In summary, the work provides a rigorous probabilistic foundation for modeling indel evolution in multiple alignment, clarifies the relationship between the objective functions used in alignment software and statistical likelihoods, and establishes conditions under which parameter estimation is provably consistent. These results bridge the gap between heuristic alignment algorithms and formal statistical inference, opening the way for future development of model‑based alignment tools and more reliable phylogenetic analyses.
Comments & Academic Discussion
Loading comments...
Leave a Comment