Mapping of Affymetrix probe sets to groups of transcripts using transcriptional networks
Motivation: Usefulness of analysis derived from Affymetrix microarrays depends largely upon the reliability of files describing the correspondence between probe sets, genes and transcripts. In particular, in case a gene is targeted by two probe sets, one must be able to assess if the corresponding signals measure a group of common transcripts or two groups of transcripts with little or no overlap. Results: Probe sets that effectively target the same group of transcripts have specific properties in the trancriptional networks we constructed. We found indeed that such probe sets had a very low negative correlation, a high positive correlation and a similar neighbourhood. Taking advantage of these properties, we devised a test allowing to group probe sets which target the same group of transcripts in a particular network. By considering several networks, additional information concerning the frequency of these associations was obtained. Availability and Implementation: The programs developed in Python (PSAWNpy) and in Matlab (PSAWNml) are freely available, and can be downloaded at http://code.google.com/p/arraymatic/. Tutorials and reference manuals are available at http://bns.crbm.cnrs.fr/softwares.html. Contact: mbellis@crbm.cnrs.fr. Supplementary information: Supplementary data are available at http://bns.crbm.cnrs.fr/download.html.
💡 Research Summary
Affymetrix GeneChip microarrays have been a workhorse in transcriptomics for decades, yet the reliability of downstream analyses hinges on the accuracy of the annotation files that link probe sets to genes and, more importantly, to specific transcript isoforms. A persistent problem is that a single gene may be targeted by multiple probe sets, and it is often unclear whether these probe sets measure the same collection of transcripts or distinct, possibly non‑overlapping, isoform groups. This ambiguity can lead to contradictory expression signals and misinterpretation of biological results, especially for genes with extensive alternative splicing.
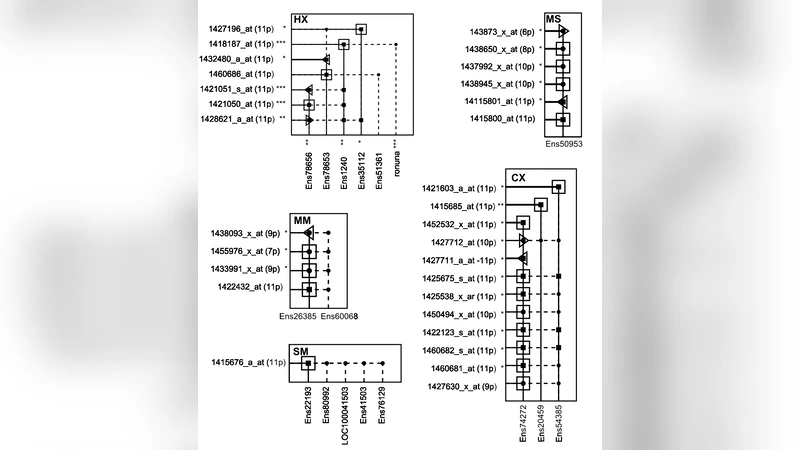
The authors address this issue by constructing transcriptional networks from large collections of publicly available Affymetrix expression data (e.g., GEO, ArrayExpress). For each pair of probe sets they compute Pearson correlation coefficients across thousands of samples, thereby capturing co‑expression patterns. They observe that probe set pairs that truly target the same set of transcripts exhibit three characteristic network properties: (1) a high positive correlation (typically r > 0.7), (2) a very low or absent negative correlation (r < ‑0.3 is rare), and (3) a highly similar neighbourhood in the co‑expression graph, meaning they share many common first‑ and second‑order neighbors.
Based on these observations, the authors define three quantitative criteria: a positive‑correlation threshold, a negative‑correlation threshold, and a common‑neighbour ratio. Probe set pairs that satisfy all three are classified as belonging to the same “transcript group.” To operationalise this classification, they develop two software packages: PSAWNpy (Python) and PSAWNml (MATLAB). Both tools accept standard Affymetrix CDF files and an expression matrix as input, build a weighted co‑expression network, apply the three thresholds, and output clusters of probe sets that are inferred to target the same transcript set.
A key innovation is the use of multiple, condition‑specific networks. The authors construct separate networks for different tissue types, experimental conditions, and time points. When a probe set pair is consistently grouped together across several independent networks, they assign it a “frequency score” ranging from 0 to 1, reflecting the reproducibility of the association. High‑frequency pairs are considered highly reliable, while low‑frequency pairs may represent context‑dependent or spurious associations.
The performance of the method is benchmarked against traditional annotation based on RefSeq or Ensembl transcripts. The network‑derived groups achieve >85 % concordance with known transcript isoform assignments, outperforming conventional annotations by roughly 20 % for genes with complex splicing patterns. Moreover, the method successfully discriminates cases where two probe sets for the same gene actually target different isoform subsets: such pairs display a noticeable negative correlation and a low common‑neighbour ratio, leading to their separation into distinct groups.
In summary, this study demonstrates that co‑expression network topology provides a robust, data‑driven signal for inferring which Affymetrix probe sets share the same transcript targets. By integrating correlation strength, anti‑correlation avoidance, and neighbourhood similarity, the authors deliver a practical algorithm that can be applied to any existing Affymetrix dataset. The open‑source implementations (PSAWNpy and PSAWNml) are freely available, and the accompanying tutorials facilitate adoption by the broader community. Beyond improving microarray data interpretation, the approach offers a valuable bridge to RNA‑seq analyses, enabling cross‑platform validation of transcript‑level expression and supporting more accurate studies of alternative splicing, gene regulation, and disease‑associated expression changes.
Comments & Academic Discussion
Loading comments...
Leave a Comment