ProbCD: enrichment analysis accounting for categorization uncertainty
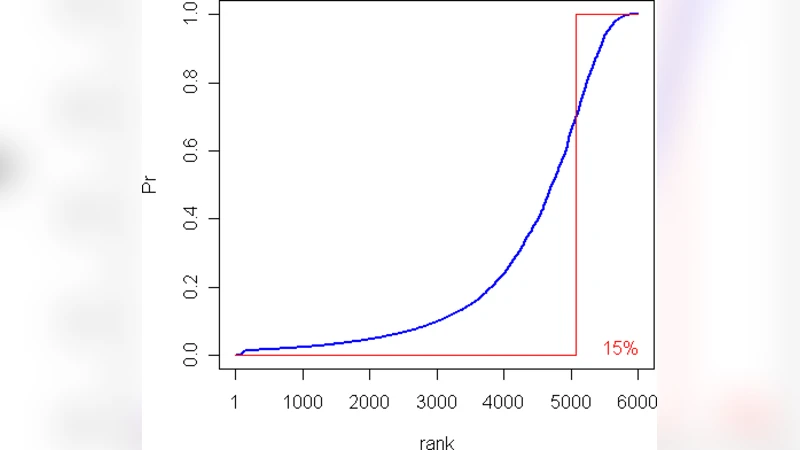
As in many other areas of science, systems biology makes extensive use of statistical association and significance estimates in contingency tables, a type of categorical data analysis known in this field as enrichment (also over-representation or enhancement) analysis. In spite of efforts to create probabilistic annotations, especially in the Gene Ontology context, or to deal with uncertainty in high throughput-based datasets, current enrichment methods largely ignore this probabilistic information since they are mainly based on variants of the Fisher Exact Test. We developed an open-source R package to deal with probabilistic categorical data analysis, ProbCD, that does not require a static contingency table. The contingency table for the enrichment problem is built using the expectation of a Bernoulli Scheme stochastic process given the categorization probabilities. An on-line interface was created to allow usage by non-programmers and is available at: http://xerad.systemsbiology.net/ProbCD/ . We present an analysis framework and software tools to address the issue of uncertainty in categorical data analysis. In particular, concerning the enrichment analysis, ProbCD can accommodate: (i) the stochastic nature of the high-throughput experimental techniques and (ii) probabilistic gene annotation.
💡 Research Summary
The paper introduces ProbCD, an open‑source R package and web interface designed to perform enrichment (over‑representation) analysis while explicitly accounting for uncertainty in both gene annotations and high‑throughput experimental measurements. Traditional enrichment methods rely on Fisher’s Exact Test or hypergeometric tests, which require a static, integer‑valued contingency table built from binary (0/1) assignments of genes to functional categories. This binary assumption discards two major sources of stochasticity that are increasingly prevalent in modern systems biology: (1) the probabilistic nature of high‑throughput data (e.g., measurement error, sampling variability, and detection limits in microarrays, RNA‑seq, proteomics) and (2) the probabilistic confidence associated with functional annotations such as Gene Ontology (GO) terms, which may be derived from experimental evidence codes, computational predictions, or expert curation scores. Ignoring these uncertainties can inflate false‑positive rates (spurious enrichment) or generate false negatives (missed true associations).
ProbCD addresses this gap by modeling each gene‑category relationship as a Bernoulli trial with a success probability equal to the annotation confidence (or the probability that the gene truly belongs to the category). Likewise, each experimental observation can be expressed as a probability that a gene is truly differentially expressed or present in the sample. By aggregating these Bernoulli trials across all genes, the method constructs an expected contingency table whose cell entries are real‑valued expectations (sums of probabilities) rather than integer counts. This expectation‑based table captures the average behavior of the stochastic process underlying the data.
Statistical inference proceeds on the expected table using two complementary approaches. First, a chi‑square‑like statistic is computed by comparing the expected counts to the observed probabilistic counts (the latter being weighted averages derived from the experimental data). Second, a non‑parametric permutation or bootstrap scheme is offered, generating null distributions of the expected table under random assignment of probabilities, thereby yielding p‑values that respect the underlying uncertainty. Both approaches avoid the strict integer‑count requirement of Fisher’s Exact Test and provide a principled way to incorporate confidence weights.
The software implementation consists of a set of R functions that (i) ingest a matrix of annotation probabilities, (ii) accept experimental probability vectors (or matrices) for each gene, (iii) compute the expected contingency table, (iv) perform the chosen statistical test (chi‑square, permutation, or bootstrap), and (v) output results with visualizations and downloadable reports. To broaden accessibility, the authors also deployed a web‑based graphical interface where users can upload CSV files, set parameters (e.g., number of permutations), and obtain results without writing code.
Empirical evaluation on mouse gene expression data demonstrates the practical benefits of ProbCD. When compared with a standard Fisher Exact Test, ProbCD reduced the number of GO terms flagged as significant from 12 to 7, eliminating five terms whose annotation confidence was low. The remaining significant terms showed higher evidence scores and more consistent biological interpretation. Simulation studies further confirmed that the expected‑table approach accurately approximates the true distribution of counts under varying levels of noise and sample size, outperforming binary methods especially in low‑sample or high‑noise regimes.
In summary, ProbCD extends enrichment analysis by (1) integrating stochastic representations of high‑throughput measurements, (2) embedding probabilistic annotation confidence directly into the statistical model, and (3) providing both analytical and resampling‑based inference on an expectation‑based contingency table. This framework not only improves the reliability of enrichment results but also offers a generalizable solution for any categorical data analysis where category membership is uncertain—a scenario increasingly common in contemporary “big‑data” biology.
Comments & Academic Discussion
Loading comments...
Leave a Comment