A novel method for identification of local conformational changes in proteins
Motivation: Proteins are known to undergo conformational changes in the course of their functions. The changes in conformation are often attributable to a small fraction of residues within the protein. Therefore identification of these variable regions is important for an understanding of protein function. Results: We propose a novel method for identification of local conformational changes in proteins. In our method, backbone conformations are encoded into a sequence of letters from a 16-letter alphabet (called D2 codes) to perform structural comparison. Since we do not use clustering analysis to encode local structures, the D2 codes not only provides a intuitively understandable description of protein structures, but also covers wide varieties of distortions. This paper shows that the D2 codes are better correlated with changes in the dihedral angles than a structural alphabet and a secondary structure description. In the case of the N37S mutant of HIV-1 protease, local conformational changes were captured by the D2 coding method more accurately than other methods. The D2 coding also provided a reliable representation of the difference between NMR models of an HIV-1 protease mutant.
💡 Research Summary
The paper introduces a novel computational framework, termed D2 coding, for detecting local conformational changes in proteins. Traditional approaches—such as monitoring backbone dihedral angles (ϕ/ψ), Cα‑Cα distances, or using structural alphabets derived from clustering—often miss subtle variations because they are either sensitive to experimental noise, rely on coarse-grained representations, or cannot capture changes that do not affect secondary‑structure assignments.
D2 coding converts a protein’s three‑dimensional backbone into a one‑dimensional string of characters drawn from a 16‑letter alphabet. The conversion proceeds in three steps. First, the protein is divided into overlapping five‑residue fragments. Each fragment is approximated by a sequence of five tetrahedra that fill space; each tetrahedron consists of four short edges and two long edges with a fixed length ratio (√3/2). Adjacent tetrahedra share a face, forming a “folded tetrahedron sequence.” Second, the algorithm defines a “gradient” for each tetrahedron as the direction of the edge that is not shared with its predecessor. When the gradient of a tetrahedron differs from that of the previous one, a binary digit ‘1’ is recorded; otherwise ‘0’ is recorded. This yields a 0/1 string of length five for each fragment. Third, the five‑bit binary word is mapped onto a base‑32 symbol (0‑9, A‑V), and the symbols are placed at the central Cα atom of the fragment. Concatenating these symbols along the protein chain produces the D2 code, a compact representation that directly reflects geometric changes without any clustering step.
Because the encoding is deterministic and based on actual geometry, D2 codes capture a wide variety of local distortions—including bends, twists, and subtle shifts—while deliberately excluding pathological “U‑turns” that would otherwise confound the representation. Variable regions are identified by comparing D2 codes across multiple experimental models of the same protein (different crystal structures, NMR ensembles, or mutants). A residue that assumes more than one D2 symbol across the models is flagged as conformationally variable; the most frequent symbol is designated the “major” code, while the others are “minor.”
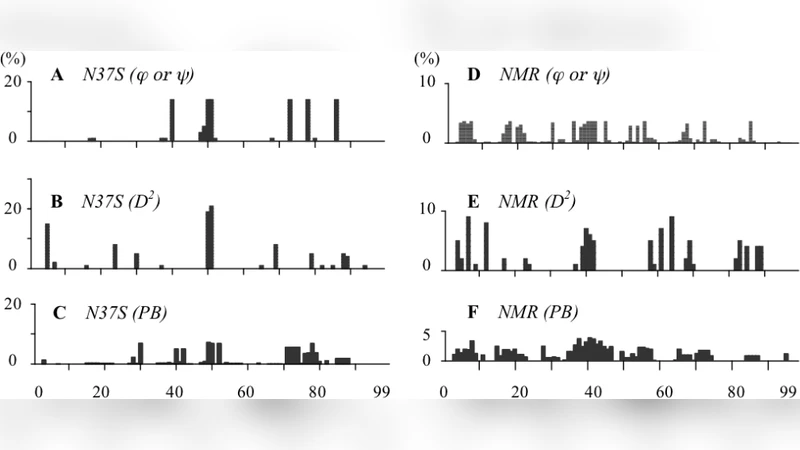
The authors benchmarked D2 coding against three established methods: PB coding (a structural alphabet based on backbone fragments), DSSP secondary‑structure assignment, and a simple ϕ/ψ angle‑difference approach. Performance was evaluated on three gold‑standard definitions derived from dihedral‑angle changes: (1) ANGL – residues with either ϕ or ψ differing by ≥30°, (2) ANGL_SUPP – any residue showing any angle change, and (3) ANGL_CORE – residues with extreme changes (≥100° in one angle or ≥30° in both). Using a dataset of 60 proteins with multiple structures, 72 crystal structures of the HIV‑1 protease N37S mutant, and 28 NMR models of another protease mutant, the authors computed accuracy, sensitivity, specificity, and Matthews correlation coefficient (MCC).
Results show that D2 coding achieves the highest accuracy and MCC for the ANGL region, indicating a balanced ability to detect true positives while limiting false positives. For ANGL_SUPP, D2 coding attains the highest specificity, meaning it rarely flags a residue as variable when it is not. PB coding, by contrast, is most sensitive for the ANGL_CORE region, capturing large dihedral swings but at the cost of more false positives. Overall, D2 coding outperforms the other methods in detecting moderate, biologically relevant conformational changes.
A particularly compelling case study involves the N37S HIV‑1 protease mutant. Across 72 X‑ray structures, D2 coding precisely identified the mutation site and adjacent loop regions that undergo subtle rearrangements, whereas PB and DSSP codes either missed these changes or over‑generalized them. In the NMR ensemble of 28 models, D2 coding successfully highlighted residues with multiple symbols, reflecting the intrinsic flexibility of the protein in solution—a scenario where traditional angle‑based metrics are less informative.
Methodologically, D2 coding offers practical advantages: the resulting string can be aligned with standard sequence‑alignment tools, eliminating the need for specialized 3‑D superposition algorithms. The approach is computationally lightweight for typical protein sizes, though the initial tetrahedron placement and gradient calculation involve rotation and translation steps that may become costly for very large complexes.
In conclusion, the D2 coding scheme provides an intuitive, geometry‑driven representation of protein backbones that captures local conformational variability more faithfully than existing structural alphabets or secondary‑structure descriptors. Its ability to detect subtle, functionally important changes without extensive preprocessing makes it a valuable addition to the toolbox of structural bioinformatics, with potential extensions into machine‑learning‑based prediction of mutation effects and large‑scale comparative analyses of protein families.
Comments & Academic Discussion
Loading comments...
Leave a Comment