Automated RNA structure prediction uncovers a missing link in double glycine riboswitches
The tertiary structures of functional RNA molecules remain difficult to decipher. A new generation of automated RNA structure prediction methods may help address these challenges but have not yet been experimentally validated. Here we apply four prediction tools to a remarkable class of double glycine riboswitches that exhibit ligand-binding cooperativity. A novel method (BPPalign), RMdetect, JAR3D, and Rosetta 3D modeling give consistent predictions for a new stem P0 and kink-turn motif. These elements structure the linker between the RNAs’ double aptamers. Chemical mapping on the F. nucleatum riboswitch with SHAPE, DMS, and CMCT probing, mutate-and-map studies, and mutation/rescue experiments all provide strong evidence for the structured linker. Under solution conditions that separate two glycine binding transitions, disrupting this helix-junction-helix structure gives 120-fold and 6- to 30-fold poorer association constants for the two transitions, corresponding to an overall energetic impact of 4.3 \pm 0.5 kcal/mol. Prior biochemical and crystallography studies from several labs did not include this critical element due to over-truncation of the RNA. We argue that several further undiscovered elements are likely to exist in the flanking regions of this and other RNA switches, and automated prediction tools can now play a powerful role in their detection and dissection.
💡 Research Summary
This paper presents a comprehensive validation of modern automated RNA tertiary‑structure prediction tools by applying them to double‑glycine riboswitches, a class of regulatory RNAs that display cooperative binding of two glycine molecules. The authors first assembled an expanded multiple sequence alignment of 360 riboswitch homologs, extending each sequence by 100 nucleotides into the 5′ and 3′ flanking regions that had previously been omitted from most experimental constructs. Four independent computational pipelines were then employed: a newly developed BPPalign algorithm that averages base‑pairing probabilities across homologs, the motif‑search program RMdetect, the structural database query server JAR3D, and Rosetta/FARFAR 3‑D modeling.
BPPalign identified a previously unannotated stem, termed P0, formed by nine nucleotides immediately upstream of the canonical aptamer linker. RMdetect and JAR3D both reported a high‑confidence kink‑turn (k‑turn) motif spanning the junction between P0 and the first aptamer stem (P1), with the k‑turn’s characteristic three‑residue bulge and conserved sequence pattern. Rosetta modeling inserted this k‑turn into the existing crystal structure of the Fusobacterium nucleatum riboswitch, producing a continuous helical interface without steric clashes. Notably, the modeled backbone trajectory of the k‑turn matched the electron density observed in the deposited crystal coordinates (≈4.4 Å C4′ RMSD), despite the original structure lacking explicit base‑pairing partners for this region.
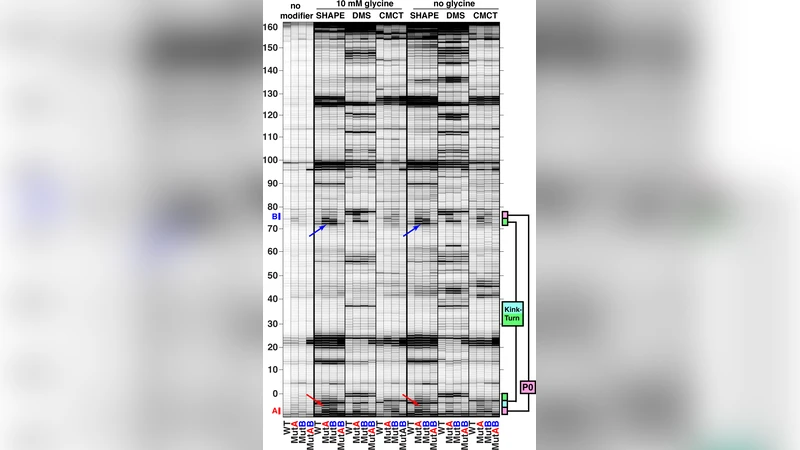
To test these predictions experimentally, the authors synthesized a minimal riboswitch construct (FN‑KTtest) that retained the natural 5′ extension. They performed nucleotide‑resolution chemical probing using SHAPE, dimethyl sulfate (DMS), and CMCT, observing protection patterns in the predicted P0‑k‑turn region that were consistent with a structured element. Mutate‑and‑map experiments introduced two single‑mutant variants (MutA disrupting the 5′ side of P0, MutB disrupting the 3′ side). Both mutants displayed increased reactivity at the disrupted base pairs and at the adjacent P1 purine‑purine stack, indicating loss of base‑pairing and helix stability. A double mutant (MutAB) that restored Watson‑Crick complementarity rescued the SHAPE, DMS, and CMCT profiles to wild‑type levels, confirming that the observed changes were due to specific base‑pair disruption rather than global folding defects.
The functional impact of the P0‑k‑turn was quantified by measuring glycine‑dependent conformational changes using DMS probing under conditions that allowed resolution of two distinct binding transitions (K1 and K2). In the wild‑type construct, the first transition had K1 ≈ 9 µM and the second K2 ≈ 1.8 mM. Both MutA and MutB required dramatically higher glycine concentrations: K1 increased ~120‑fold (≈1.1 mM) and K2 increased 6‑ to 30‑fold (10–50 mM). Thermodynamic analysis attributed these shifts to destabilization of the first binding event by 2.8 ± 0.1 kcal mol⁻¹ and of the second by an additional 1.5 ± 0.5 kcal mol⁻¹. The double mutant MutAB restored the binding constants to near‑wild‑type values (K1 ≈ 10 µM, K2 ≈ 1.3 mM), confirming that the energetic penalties arise specifically from loss of the P0‑k‑turn architecture.
These findings demonstrate that the previously overlooked P0 stem and kink‑turn motif are integral to the structural integrity and ligand‑binding energetics of double‑glycine riboswitches. The work also highlights a broader methodological lesson: automated RNA structure prediction tools, when combined with rigorous biochemical validation, can uncover functional elements hidden in flanking regions that are routinely truncated in traditional studies. The authors suggest that additional, as yet unidentified motifs may reside further outward in riboswitch sequences, influencing regulation in ways not captured by current models. This study thus establishes a powerful pipeline for discovering and characterizing cryptic RNA structural features, with implications for the design of synthetic riboswitches, interpretation of chemogenomic data, and improvement of crystallographic constructs for other regulatory RNAs.
Comments & Academic Discussion
Loading comments...
Leave a Comment