Thermodynamics of polymer adsorption to a flexible membrane
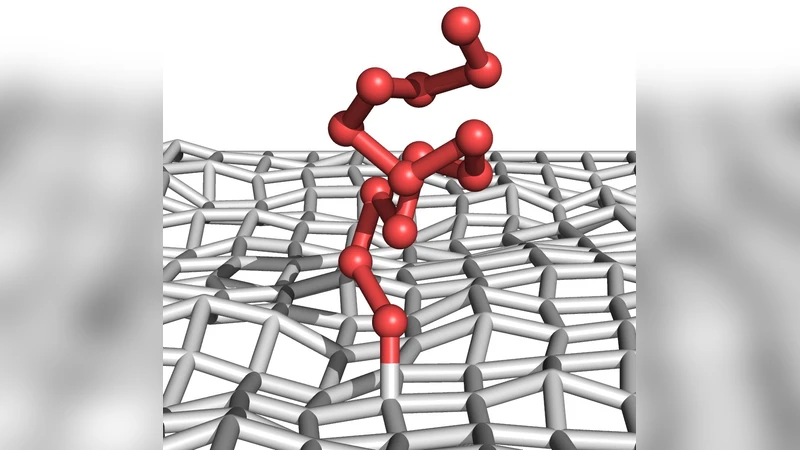
We analyze the structural behavior of a single polymer chain grafted to an attractive, flexible surface. Our model is composed of a coarse-grained bead-and-spring polymer and a tethered membrane. By means of extensive parallel tempering Monte Carlo simulations it is shown that the system exhibits a rich phase behavior ranging from highly ordered, compact to extended random coil structures and from desorbed to completely adsorbed or even partially embedded conformations. These findings are summarized in a pseudophase diagram indicating the predominant class of conformations as a function of the external parameters temperature and polymer-membrane interaction strength. By comparison with adsorption to a stiff membrane surface it is shown that the flexibility of the membrane gives rise to qualitatively new behavior such as stretching of adsorbed conformations.
💡 Research Summary
This paper investigates the structural and thermodynamic behavior of a single polymer chain grafted to an attractive, flexible membrane using a coarse‑grained off‑lattice bead‑and‑spring model combined with parallel tempering Monte Carlo simulations. The polymer consists of N = 13 monomers that interact via a Lennard‑Jones (LJ) potential for non‑bonded pairs and a finitely extensible nonlinear elastic (FENE) potential for bonded neighbors. The membrane is represented by a square lattice of Lx = Ly = 27 nodes, each connected to its nearest neighbors by identical FENE springs and prevented from overlapping by a hard‑sphere repulsion. Polymer–membrane interactions are also modeled by an LJ potential, with the interaction strength ε_pm serving as an external control parameter varied between 0.05 and 1.50. One polymer end is anchored to the central membrane node by a FENE bond, allowing the chain to explore all conformations while remaining tethered.
Two scenarios are compared: (i) a “stiff” membrane where all nodes are fixed to their ground‑state lattice positions, and (ii) a “flexible” membrane that can thermally fluctuate around the planar ground state. Parallel tempering (PT) simulations were performed with 24 replicas spanning temperatures from T = 0.021 to 1.5, each replica undergoing 8 × 10⁶ Monte‑Carlo sweeps; exchanges were attempted every 20 sweeps. Observables include total energy and its components, heat capacities, radius of gyration (overall, parallel, and perpendicular components), the center‑of‑mass height above the membrane, and contact numbers (polymer‑polymer and polymer‑membrane) defined via an energy threshold.
The results are summarized in pseudophase diagrams in the (ε_pm, T) plane. For the stiff membrane, seven distinct structural regimes are identified: (a) Desorbed Compact (DC) – icosahedral, highly ordered compact structures attached to the surface; (b) Globular (G) – compact but disordered globules; (c) Desorbed Expanded (DE) – random‑coil conformations confined to the half‑space above the membrane; (d) Adsorbed Compact Single‑Layer (AC1) – flat, disk‑like compact films lying on the lattice; (e) Adsorbed Compact Double‑Layer (AC2) – a bottom layer adsorbed to the membrane with a second semi‑spherical droplet on top; (f) Adsorbed Expanded Double‑Layer (AE2) – partially adsorbed coils extending into the third dimension; and (g) Adsorbed Expanded Single‑Layer (AE1) – fully adsorbed, two‑dimensional random coils. Transitions between these regimes are detected via peaks in heat capacity, abrupt changes in contact numbers, and variations in the sphericity ratio Ψ_r = √2 R_g,⊥ / R_g,‖.
When the membrane is allowed to fluctuate, the overall topology of the diagram remains similar, but new qualitative features emerge. The flexible membrane can locally deform to accommodate polymer monomers, leading to “partial embedding” where the polymer penetrates the membrane surface. This flexibility induces a stretching effect: for comparable ε_pm, the perpendicular component of the radius of gyration (R_g,⊥) is larger than in the stiff case, and the number of polymer‑membrane contacts shows a non‑monotonic behavior (increase in AC2, decrease in AE2). Consequently, the AC2 ↔ AE2 transition becomes smoother, and the membrane’s elastic response creates additional intermediate states not present for a rigid substrate.
Key insights from the study are: (1) Membrane flexibility fundamentally alters the adsorption pathway, generating novel conformational phases such as partially embedded droplets and stretched adsorbed coils. (2) Contact numbers and the parallel/perpendicular gyration components serve as sensitive order parameters for detecting adsorption, compaction, and stretching transitions. (3) Parallel tempering combined with multiple‑histogram reweighting provides an efficient route to explore the rugged energy landscape of polymer‑membrane systems, even for relatively short chains. (4) The coarse‑grained model captures essential physics relevant to biological scenarios where polymers (e.g., proteins, peptides) interact with fluctuating lipid bilayers, offering a tractable framework for future extensions that could include membrane tension, curvature, or heterogeneous interaction sites.
In conclusion, the paper demonstrates that the thermodynamic phase behavior of polymer adsorption is markedly richer when the substrate is flexible. The presented pseudophase diagrams and the identified order parameters lay the groundwork for systematic studies of more realistic biomembrane‑polymer interactions and may guide experimental investigations of polymer adsorption on soft interfaces.
Comments & Academic Discussion
Loading comments...
Leave a Comment