A brief overview on the BioPAX and SBML standards for formal presentation of complex biological knowledge
A brief informal overview on the BioPAX and SBML standards for formal presentation of complex biological knowledge.
💡 Research Summary
Modern biology is increasingly driven by high‑throughput technologies that generate massive, structured datasets describing cellular mechanisms. To transform these data into usable knowledge, standardized, computer‑readable representations are essential. This paper provides a concise yet comprehensive overview of two widely adopted XML‑based standards for representing biological network information: BioPAX (Biological Pathway Exchange) and SBML (Systems Biology Markup Language).
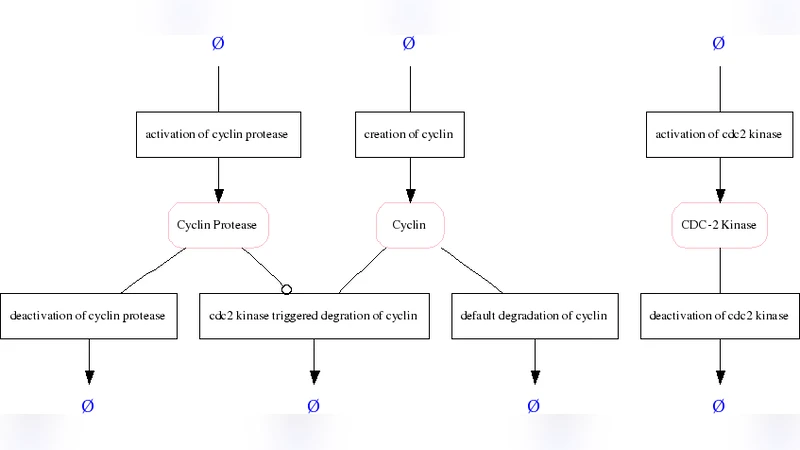
The author begins by outlining the need for formal standards, noting that traditional two‑dimensional pathway diagrams are insufficient for large‑scale computational analyses. An illustrative example—a minimal cascade model of the mitotic oscillator involving cyclin and Cdc2 kinase from the BioModels database—is used to demonstrate how even a simple network requires detailed encoding of entities, interactions, and contextual information.
BioPAX is described as an ontology‑driven framework that models biological knowledge through a class hierarchy. Core classes such as PhysicalEntity, Interaction, and Pathway are further specialized into subclasses (e.g., Protein, SmallMolecule, Conversion, Transport). This hierarchical structure enables precise annotation of molecular participants, interaction types, and subcellular locations, while also supporting hyperlinks to external resources. BioPAX Level 2 already covers metabolic and molecular interaction networks, with future extensions planned for gene regulation and signaling. The standard is supported by major pathway databases (Reactome, PID, BioCyc) and a suite of tools: Protege for graphical editing, the Rredland package for low‑level querying within the R/Bioconductor environment, and numerous open‑source analysis utilities. Importantly, BioPAX focuses on static representations; it does not encode kinetic equations or dynamic behavior.
SBML, by contrast, is built around the explicit mathematical description of biochemical reaction systems. An SBML model consists of Species (chemical entities), Compartments (spatial contexts), Reactions (with reactants, products, modifiers), and Parameters (rate constants, initial concentrations). Kinetic laws are expressed using MathML, allowing direct simulation of time‑course dynamics. SBML Level 2 supports quantitative modeling of metabolic pathways, signaling cascades, and gene regulatory networks, and integrates with standards such as MathML and CellML. The ecosystem includes over a hundred software packages for simulation (COPASI, libSBML, CellDesigner) and analysis (RSBML for R, Java libraries). While SBML provides rich dynamic capabilities, its representation of complex molecular assemblies and hierarchical relationships is less detailed than BioPAX; such features are slated for future Level 3 extensions.
The discussion compares the two standards, emphasizing their complementary nature. BioPAX excels at capturing detailed static network topology and metadata, facilitating pathway queries, cross‑database integration, and high‑level reasoning. SBML excels at quantitative simulation, enabling researchers to test hypotheses about kinetic behavior, dose‑response, and system stability. The author notes that many databases (e.g., BioModels) distribute the same network in both formats, allowing users to choose the appropriate representation for their task. A typical workflow might involve using BioPAX to retrieve and curate the structural network, then converting it to SBML for dynamic simulation.
In conclusion, BioPAX and SBML together provide a robust foundation for representing complex biological knowledge. BioPAX offers a hierarchical, ontology‑based view suitable for static pathway analysis and data integration, while SBML supplies a mathematically rigorous framework for dynamic modeling and simulation. Their interoperability and extensive tool support make them indispensable for modern systems biology, and their continued development promises even tighter integration in the future.
Comments & Academic Discussion
Loading comments...
Leave a Comment