Reachability in Biochemical Dynamical Systems by Quantitative Discrete Approximation (extended abstract)
In this paper, a novel computational technique for finite discrete approximation of continuous dynamical systems suitable for a significant class of biochemical dynamical systems is introduced. The method is parameterized in order to affect the imposed level of approximation provided that with increasing parameter value the approximation converges to the original continuous system. By employing this approximation technique, we present algorithms solving the reachability problem for biochemical dynamical systems. The presented method and algorithms are evaluated on several exemplary biological models and on a real case study.
💡 Research Summary
The paper introduces a novel quantitative discrete approximation technique for analyzing reachability in biochemical dynamical systems that are modeled by autonomous ordinary differential equations (ODEs) with multi‑affine right‑hand sides. Traditional rectangular abstraction methods partition the continuous state space into hyper‑rectangles and over‑approximate transitions between them, which often generates a large number of spurious trajectories that do not correspond to any real solution of the original ODE. To overcome this limitation, the authors propose the Quantitative Discrete Approximation Automaton (QD‑AA).
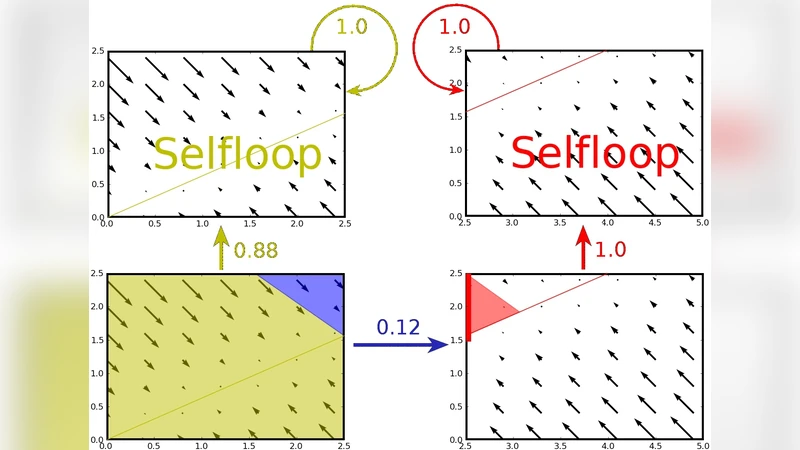
The method proceeds as follows: (1) a non‑uniform rectangular grid is defined by a set of thresholds for each variable, producing a finite collection of rectangles; (2) for each rectangle, each facet (entry face) is discretized into small “entry sets” using a user‑controlled granularity parameter κ; (3) local numerical simulations are performed from sample points inside each entry set to determine which exit facet the trajectories reach; (4) subsets of the entry set that all flow to the same exit facet are identified as focal subsets; (5) a transition weight is assigned to each entry‑exit pair, defined as the (n‑1)-dimensional Lebesgue measure of the focal subset divided by the measure of the whole entry set. These weighted transitions form a finite‑state, discrete‑time Markov chain—the QD‑AA.
Two central theorems are proved. Theorem 3.2 shows that the QD‑AA correctly captures the behavior of the original continuous system in the sense of a probabilistic abstraction, while Theorem 3.3 establishes convergence: as κ → ∞ (i.e., the facet discretization becomes arbitrarily fine), the transition weights converge to the true Lebesgue measures of the corresponding continuous regions, and thus the QD‑AA’s reachability results converge to those of the original ODE system.
An algorithmic pipeline is presented: (i) generate the rectangular partition; (ii) for each rectangle and each facet, sample entry points according to κ; (iii) integrate the ODE locally (e.g., using Runge‑Kutta) to identify exit facets; (iv) compute transition probabilities; (v) perform standard Markov‑chain reachability analysis (e.g., matrix exponentiation or iterative probability propagation) to answer questions such as “what is the probability that a species concentration exceeds a given threshold starting from a set of initial conditions?”.
The authors evaluate the approach on three case studies: a simple 2‑dimensional linear system, a multi‑affine non‑linear system, and a realistic Escherichia coli metabolic network (7 species, 12 reactions). Compared with classical rectangular abstraction, QD‑AA dramatically reduces spurious transitions (from ~30 % to <5 % in the non‑linear case) and yields accurate quantitative estimates of minimal and maximal reachable concentrations. In the E. coli model, the probability of reaching a target concentration region computed by QD‑AA (0.92) matches Monte‑Carlo simulation results within an error of 0.03, while requiring far fewer simulation runs.
Complexity analysis shows that the dominant cost lies in the facet‑wise sampling phase, which scales as O(|Rect| · n · κ^{n‑1} · T), where T is the cost of a single ODE integration step. Nevertheless, for typical biochemical models (dimensions up to 10) and moderate κ values, the method runs comfortably on modern multi‑core hardware.
In conclusion, the paper provides a bridge between formal verification techniques and numerical simulation for biochemical systems. By augmenting rectangular abstraction with locally measured transition probabilities, the QD‑AA offers a tunable trade‑off between computational effort and approximation accuracy, eliminating much of the conservatism inherent in previous over‑approximations while preserving scalability. Future work is suggested on extending the framework to non‑multi‑affine dynamics, adaptive grid refinement, and stochastic reaction networks.
Comments & Academic Discussion
Loading comments...
Leave a Comment