Reaction-Diffusion-Delay Model for EPO/TNF-$alpha$? Interaction in Articular Cartilage Lesion Abatement
Injuries to articular cartilage result in the development of lesions that form on the surface of the cartilage. Such lesions are associated with articular cartilage degeneration and osteoarthritis. The typical injury response often causes collateral damage, primarily an effect of inflammation, which results in the spread of lesions beyond the region where the initial injury occurs. We present a minimal mathematical model based on known mechanisms to investigate the spread and abatement of such lesions. In particular we represent the “balancing act” between pro-inflammatory and anti-inflammatory cytokines that is hypothesized to be a principal mechanism in the expansion properties of cartilage damage during the typical injury response. We present preliminary results of in vitro studies that confirm the anti-inflammatory activities of the cytokine erythropoietin (EPO). We assume that the diffusion of cytokines determine the spatial behaviour of injury response and lesion expansion so that a reaction-diffusion system involving chemical species and chondrocyte cell state population densities is a natural way to represent cartilage injury response. We present computational results using the mathematical model showing that our representation is successful in capturing much of the interesting spatial behaviour of injury associated lesion development and abatement in articular cartilage. Further, we discuss the use of this model to study the possibility of using EPO as a therapy for reducing the amount of inflammation induced collateral damage to cartilage during the typical injury response. The mathematical model presented herein suggests that not only are anti-inflammatory cytokines, such as EPO necessary to prevent chondrocytes signaled by pro-inflammatory cytokines from entering apoptosis, they may also influence how chondrocytes respond to signaling by pro-inflammatory cytokines.
💡 Research Summary
**
This paper investigates the spatial dynamics of cartilage lesion expansion and its possible abatement through the interaction of pro‑inflammatory (TNF‑α) and anti‑inflammatory (erythropoietin, EPO) cytokines. The authors develop a minimal reaction‑diffusion‑delay (RDD) model that captures the essential biochemical and cellular processes observed in vitro and in vivo after an articular cartilage injury.
Biological background – An acute injury causes necrotic chondrocytes to release damage‑associated molecular patterns (DAMPs). DAMPs stimulate nearby healthy chondrocytes to enter a catabolic state (S_T), where they produce TNF‑α and reactive oxygen species (ROS). TNF‑α further drives catabolic cells to express the erythropoietin receptor (EPOR), creating an EPOR‑active subpopulation (S_A). ROS, after a 20–24 h delay, triggers healthy cells to synthesize EPO. EPO, in turn, can rescue EPOR‑active cells back to the healthy state, but TNF‑α suppresses EPO production, establishing a competitive balance that determines whether the lesion expands or contracts.
Mathematical formulation – The model consists of five partial differential equations (PDEs) for the concentrations of ROS (R), DAMPs (M), TNF‑α (F), EPO (P), and extracellular matrix degradation (U). Diffusion terms (D_R∇²R, etc.) describe spatial spread, while production terms (σ_R S_T, σ_F S_T, etc.) link chemical generation to specific cell states. Natural decay is modeled by linear loss terms (δ_R R, …). Four ordinary differential equations (ODEs) describe the dynamics of cell densities: healthy (C), catabolic (S_T), EPOR‑active (S_A), and necrotic (D_N). Transitions between states are governed by Michaelis‑Menten‑like terms multiplied by a Heaviside‑type switch H(s) that becomes zero when EPO exceeds a critical concentration P_c, thereby limiting the pro‑inflammatory drive. Two explicit time delays are incorporated: τ₁ (8–12 h) for EPOR expression after TNF‑α signaling, and τ₂ (20–24 h) for EPO synthesis after ROS exposure.
Simulation scenarios – The authors explore two contrasting parameter sets. (1) No EPO therapy: The positive feedback loop of DAMPs → TNF‑α → catabolic cells → more DAMPs remains unchecked (H = 1). The lesion expands radially, EPOR‑active cells accumulate, and a large fraction of chondrocytes undergo apoptosis, mimicking uncontrolled inflammation. (2) Early EPO administration: An exogenous EPO pulse is introduced shortly after injury. The switch H(s) turns off once P > P_c, suppressing further DAMP‑ and TNF‑α‑driven transitions. EPOR‑active cells revert to the healthy state, the lesion boundary stabilizes, and overall tissue loss is dramatically reduced.
Key insights –
- Time delays are crucial: The model shows that the 8–12 h lag before EPOR expression creates a therapeutic window; delivering EPO before this window closes maximizes rescue of catabolic cells.
- Threshold behavior: The Heaviside switch captures a bistable system where a modest increase in EPO can flip the network from a runaway inflammatory state to a self‑limiting one.
- Spatial diffusion matters: By solving the PDEs, the authors demonstrate that diffusion of cytokines determines the radius of the “penumbra” of at‑risk cells, providing a mechanistic explanation for lesion spread observed clinically.
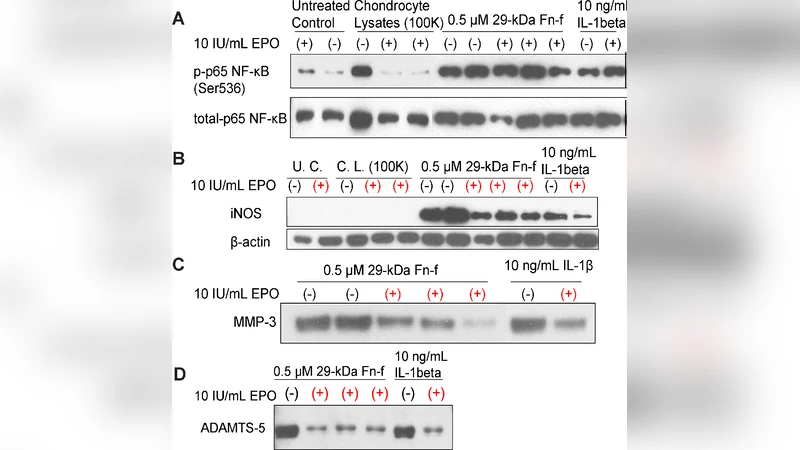
- Minimal yet predictive: Despite using only five chemicals and four cell states, the model reproduces qualitative features of in‑vitro experiments (e.g., EPO’s suppression of iNOS, MMP‑3, ADAMTS‑5) and predicts outcomes of hypothetical therapeutic regimens.
Limitations and future directions – Parameter values are largely drawn from literature averages; systematic sensitivity analysis and experimental calibration are needed. The model omits mechanical loading, vascular influx, and other cytokines such as IL‑1β, which could be incorporated in an extended framework. Extending the geometry to three‑dimensional joint surfaces and coupling to cartilage biomechanics would enhance clinical relevance. Finally, the authors suggest that the RDD framework could serve as a rapid, inexpensive screening tool for anti‑inflammatory strategies before costly animal studies.
In summary, the paper presents a thoughtfully constructed reaction‑diffusion‑delay model that links cytokine signaling, cell‑state transitions, and spatial diffusion to explain how EPO can counteract TNF‑α‑driven cartilage lesion expansion. The work bridges experimental observations with quantitative theory, offering a valuable platform for exploring therapeutic timing, dosage, and combination strategies aimed at preserving articular cartilage after injury.
Comments & Academic Discussion
Loading comments...
Leave a Comment