Review on Feature Selection Techniques and the Impact of SVM for Cancer Classification using Gene Expression Profile
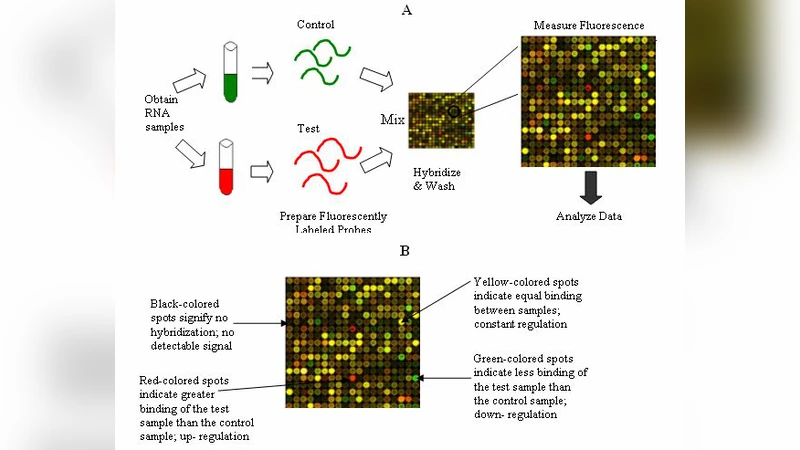
The DNA microarray technology has modernized the approach of biology research in such a way that scientists can now measure the expression levels of thousands of genes simultaneously in a single experiment. Gene expression profiles, which represent the state of a cell at a molecular level, have great potential as a medical diagnosis tool. But compared to the number of genes involved, available training data sets generally have a fairly small sample size for classification. These training data limitations constitute a challenge to certain classification methodologies. Feature selection techniques can be used to extract the marker genes which influence the classification accuracy effectively by eliminating the un wanted noisy and redundant genes This paper presents a review of feature selection techniques that have been employed in micro array data based cancer classification and also the predominant role of SVM for cancer classification.
💡 Research Summary
The reviewed paper provides a comprehensive overview of feature selection techniques applied to microarray‑based cancer classification and highlights the pivotal role of Support Vector Machines (SVM) in this domain. The authors begin by describing the rapid evolution of DNA microarray technology, which enables simultaneous measurement of expression levels for tens of thousands of genes. While this high‑throughput capability offers unprecedented insight into cellular states, it also creates a classic “large‑p, small‑n” problem: the number of features (genes) far exceeds the number of available samples. This imbalance leads to over‑fitting, inflated computational costs, and difficulty in extracting biologically meaningful signals.
To address these challenges, the paper categorizes feature selection methods into three main families: filter, wrapper, and embedded approaches. Filter methods rank individual genes using statistical or information‑theoretic criteria such as Information Gain, Signal‑to‑Noise Ratio, Euclidean distance, Pearson correlation, and Euclidean distance. Their strengths lie in computational efficiency and ease of implementation, making them suitable for very large datasets. However, because they treat each gene independently, they cannot capture inter‑gene redundancy or synergistic effects, often resulting in the selection of highly correlated, redundant markers.
Wrapper methods integrate a learning algorithm into the search process. Subsets of genes are evaluated by training a classifier (commonly SVM, k‑NN, or decision trees) and measuring cross‑validation accuracy. Meta‑heuristics such as Genetic Algorithms (GA), Particle Swarm Optimization (PSO), and Simulated Annealing are frequently employed to explore the combinatorial space. Wrappers can model feature interactions and typically achieve higher predictive performance than filters, but they are computationally intensive and prone to over‑fitting, especially when the sample size is limited.
Embedded methods fuse feature selection directly into the model training phase. The paper emphasizes SVM‑based Recursive Feature Elimination (RFE) as a prime example. In SVM‑RFE, the weight vector obtained from the optimal hyperplane is used to rank genes; those with the smallest absolute weights are iteratively removed. This process simultaneously reduces dimensionality and optimizes the classifier, leveraging the margin‑maximization principle of SVMs. Because SVMs can employ kernel functions, they handle non‑linear relationships in gene expression data effectively. The authors argue that embedded approaches strike a balance between the low computational cost of filters and the interaction awareness of wrappers, while also providing a clear link between selected features and classification performance.
Beyond feature selection, the paper discusses the broader challenges inherent to microarray analysis: experimental bias and confounding factors, lack of standardization across platforms, high dimensionality, missing values, and the need for biological interpretability. The authors note that standards such as MIAME improve reproducibility, but careful preprocessing (normalization, background correction, log‑transformation) remains essential.
In the supervised classification section, SVM is presented as the classifier of choice for cancer detection using gene expression profiles. Its structural risk minimization framework, ability to maximize the margin, and flexibility through kernel tricks give it superior generalization compared to traditional classifiers. Empirical studies cited in the review demonstrate that SVM, particularly when combined with RFE, consistently yields higher accuracy and more stable models across diverse cancer datasets. Moreover, the decision function of SVM provides a quantitative measure of class confidence, facilitating downstream biological interpretation and potential biomarker discovery.
The paper concludes by acknowledging that while filter, wrapper, and embedded methods each have merits, the integration of SVM‑based embedded selection (e.g., RFE) currently offers the most effective solution for the high‑dimensional, low‑sample microarray setting. Future research directions suggested include multi‑omics integration, pathway‑aware weighting schemes, and hybrid deep‑learning architectures that retain interpretability while exploiting large‑scale data. Overall, the review underscores that sophisticated feature selection, anchored by robust classifiers like SVM, is indispensable for translating microarray gene expression data into reliable, clinically relevant cancer diagnostics.
Comments & Academic Discussion
Loading comments...
Leave a Comment