Covalent bond symmetry breaking and protein secondary structure
Both symmetry and organized breaking of symmetry have a pivotal r^ole in our understanding of structure and pattern formation in physical systems, including the origin of mass in the Universe and the chiral structure of biological macromolecules. Here we report on a new symmetry breaking phenomenon that takes place in all biologically active proteins, thus this symmetry breaking relates to the inception of life. The unbroken symmetry determines the covalent bond geometry of a sp3 hybridized carbon atom. It dictates the tetrahedral architecture of atoms around the central carbon of an amino acid. Here we show that in a biologically active protein this symmetry becomes broken. Moreover, we show that the pattern of symmetry breaking is in a direct correspondence with the local secondary structure of the folded protein.
💡 Research Summary
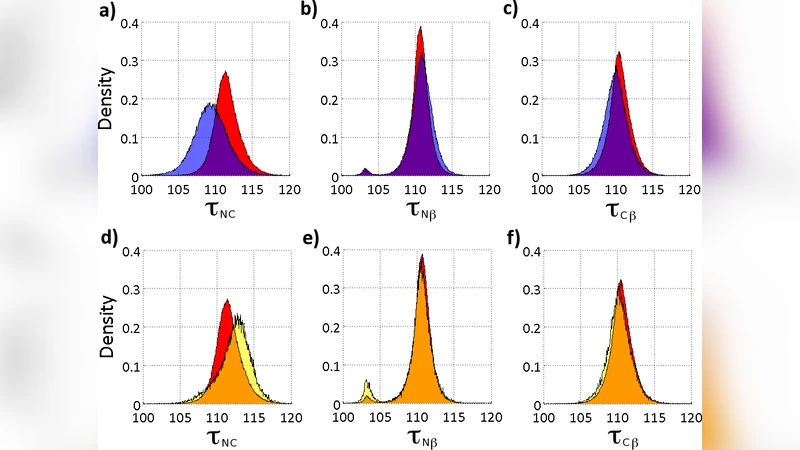
The authors report a previously unrecognized symmetry‑breaking phenomenon in the covalent geometry of the backbone Cα atom of biologically active proteins. Conventional protein modeling assumes that the Cα carbon, being sp³ hybridized, retains an ideal tetrahedral geometry independent of the protein’s secondary structure. By analysing a large set of high‑resolution (≤2 Å) protein structures from the Protein Data Bank, they find that the N‑Cα‑C bond angle (τ_NC) varies systematically with local secondary structure (α‑helix, β‑sheet, 3/10‑helix), whereas the adjacent N‑Cα‑Cβ (τ_Nβ) and C‑Cα‑Cβ (τ_Cβ) angles remain essentially constant. This indicates that the tetrahedral symmetry is broken in a manner that is fully determined by the local secondary structure, a finding that challenges the standard force‑field assumption of a fixed equilibrium angle.
To describe this effect, the authors adopt a geometric Frenet‑frame representation of the protein backbone, defining unit tangent, normal, and binormal vectors from the Cα coordinates. In this frame the backbone curvature κ_i (the angle between successive tangents) and torsion τ_i (the angle between successive binormals) become the fundamental variables. They then construct an energy function based on the integrable discrete nonlinear Schrödinger (DNLS) equation, which possesses an infinite hierarchy of conserved quantities. The proposed energy (Eq. 4) includes the usual DNLS terms plus the lowest‑order conserved momentum (b_τ) and helicity (a_τ) contributions, as well as a Proca‑type mass term. Because τ_i appears only quadratically, it can be eliminated via its equation of motion, leaving the backbone geometry fully determined by the κ_i profile.
The authors further introduce two additional energy contributions (E_θ and E_φ, Eqs. 5‑6) that depend on the spherical angles (θ_i, φ_i) describing the positions of the side‑chain Cβ atom relative to the Cα. These terms are also built from the lowest‑order DNLS conserved quantities, ensuring that the side‑chain geometry is coupled to the backbone curvature but remains independent in the latitudinal (θ) and longitudinal (φ) directions. Consequently, the Cβ “nutation” observed in the Frenet‑frame visualisations emerges naturally from the model.
To validate the approach, the authors apply it to the villin headpiece subdomain HP35 (PDB 1YRF), a 35‑residue protein containing three α‑helices separated by two loops. Using a two‑soliton solution of the DNLS equation to model the two loop regions, they solve for κ_i and τ_i, then compute θ_i and φ_i from the additional energy terms. With a set of six parameters per soliton (Table 1), they achieve an overall root‑mean‑square deviation (RMSD) of 0.39 Å for the combined Cα‑Cβ coordinates, which matches the experimental B‑factor accuracy and is comparable to state‑of‑the‑art molecular‑dynamics simulations that assume unbroken tetrahedral symmetry.
The study demonstrates that the observed τ_NC variation is not an artifact of refinement protocols but reflects a genuine physical symmetry breaking linked to secondary structure. By embedding this effect in an integrable DNLS‑based energy functional, the authors provide a compact, analytically tractable description of both backbone and side‑chain geometry that reproduces high‑resolution structural data. They argue that this symmetry breaking is fundamentally connected to protein folding: the broken tetrahedral symmetry is driven by the degenerate ground‑state structure of the DNLS equation, and the resulting soliton profiles encode the global folded shape.
The paper concludes by suggesting experimental verification of τ_NC dependence using next‑generation ultra‑high‑resolution X‑ray crystallography and quantum‑chemical calculations. It also highlights potential implications for protein‑structure prediction, force‑field development, and drug design, as incorporating symmetry‑breaking terms could improve the accuracy of computational models. The authors envision that the one‑to‑one correspondence between the deformed tetrahedral geometry and the protein fold may provide deeper insight into the physical underpinnings of life’s molecular machinery.
Comments & Academic Discussion
Loading comments...
Leave a Comment