Reachability in Biochemical Dynamical Systems by Quantitative Discrete Approximation
In this paper a novel computational technique for finite discrete approximation of continuous dynamical systems suitable for a significant class of biochemical dynamical systems is introduced. The method is parameterized in order to affect the imposed level of approximation provided that with increasing parameter value the approximation converges to the original continuous system. By employing this approximation technique, we present algorithms solving the reachability problem for biochemical dynamical systems. The presented method and algorithms are evaluated on several exemplary biological models and on a real case study. This is a full version of the paper published in the proceedings of CompMod 2011.
💡 Research Summary
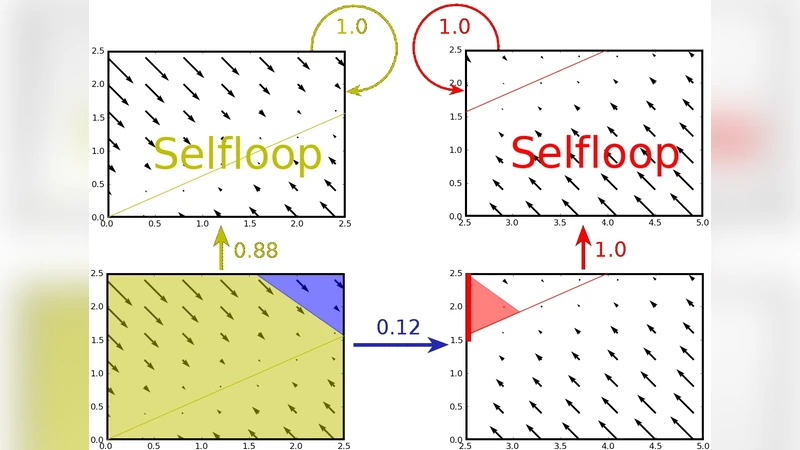
The paper introduces a novel quantitative discrete approximation (QDAA) framework for analyzing reachability in biochemical dynamical systems modeled by multi‑affine ordinary differential equations (ODEs). Traditional rectangular abstraction partitions the continuous state space into hyper‑rectangles and builds a finite transition system, but it over‑approximates transitions across rectangle facets, leading to many spurious paths that have no counterpart in the original dynamics. To mitigate this, the authors augment each rectangle with an “entry set” that records the exact portion of a facet through which trajectories actually enter the rectangle. The entry set is further decomposed into “focal subsets,” each of which consists of points whose trajectories all exit through the same facet. By measuring the (n‑1)-dimensional Lebesgue volume of each focal subset and normalizing it by the total volume of the entry set, a transition weight is obtained. These weighted transitions turn the rectangular transition system into a discrete‑time Markov chain—the QDAA—where each transition probability reflects the proportion of trajectories moving in that direction in the original continuous system.
Mathematically, the method relies on the multi‑affine nature of the vector field, which guarantees existence, uniqueness, and continuous dependence of solutions. The state space is bounded and partitioned by a (possibly non‑uniform) grid of thresholds, producing a finite set of rectangles. For a given discretization granularity parameter κ, each facet is uniformly sampled; local numerical simulations trace the flow from sampled points to identify exit facets, thereby constructing focal subsets and computing transition weights. The authors prove (Theorem 3.2) that the resulting structure is indeed a Markov chain, and (Theorem 3.3) that as κ → ∞ the QDAA converges to the exact reachable set of the continuous system.
Algorithmically, the approach consists of: (1) generating a uniform grid on each rectangle facet, (2) performing short‑time numerical integration from each grid point to determine which neighboring rectangle is reached, (3) aggregating grid points that share the same exit facet into focal subsets, and (4) assigning transition probabilities based on the relative sizes of these subsets. The global QDAA is then explored using standard graph‑search techniques (e.g., BFS/DFS) combined with probabilistic analysis to compute over‑approximations of reachable regions from a given initial set.
The methodology is evaluated on three categories of models: (i) a simple linear system, (ii) a non‑linear mass‑action kinetics model, and (iii) a realistic Escherichia coli metabolic network. In the E. coli case study, the QDAA accurately estimates maximal and minimal concentrations of a target metabolite, while dramatically reducing the number of spurious transitions compared to the classic rectangular abstraction. By varying κ, the authors demonstrate a controllable trade‑off between computational effort and approximation fidelity.
Overall, the contribution lies in providing a parameterizable, quantitatively sound discrete abstraction that bridges the gap between exhaustive formal verification and purely simulation‑based analysis for biochemical systems. The QDAA retains the scalability of rectangular abstraction while offering a principled way to quantify and diminish over‑conservatism, making it a valuable tool for systems biologists interested in global properties such as safety, invariance, and extreme concentration bounds.
Comments & Academic Discussion
Loading comments...
Leave a Comment