Population Fitness and Genetic Load of Single Nucleotide Polymorphisms Affecting mRNA splicing
Deleterious genetic variants can be evaluated as quantitative traits using information theory-based sequence analysis of recognition sites. To assess the effect of such variants, fitness and genetic load of SNPs which alter binding site affinity are derived from changes in individual information and allele frequencies. Human SNPs that alter mRNA splicing are partitioned according to their genetic load. SNPs with high genetic loads (>0.5) are common in the genome and, in many instances, predicted effects are supported by gene expression studies.
💡 Research Summary
The authors present a novel quantitative framework for assessing the functional impact of single‑nucleotide polymorphisms (SNPs) that alter mRNA splicing, by applying Shannon information theory to splice‑site recognition motifs. The central premise is that the binding affinity of a splice donor or acceptor can be expressed as an “individual information” value (Ri) measured in bits. A mutation changes this value by ΔRi; because Ri is proportional to the logarithm of the replication rate (W), the change in Ri can be translated into a change in fitness. For a biallelic SNP the fitnesses of the major (w_major) and minor (w_minor) alleles are inferred from their respective Ri values, and the population‑average fitness (Ȳ) is calculated as a frequency‑weighted sum of these fitnesses. The genetic load (L) is then defined as L = 1 − Ȳ/w_max, where w_max corresponds to the highest possible fitness (the major allele’s Ri). Assuming independence among loci, the multilocus mean fitness is the product of the individual loads.
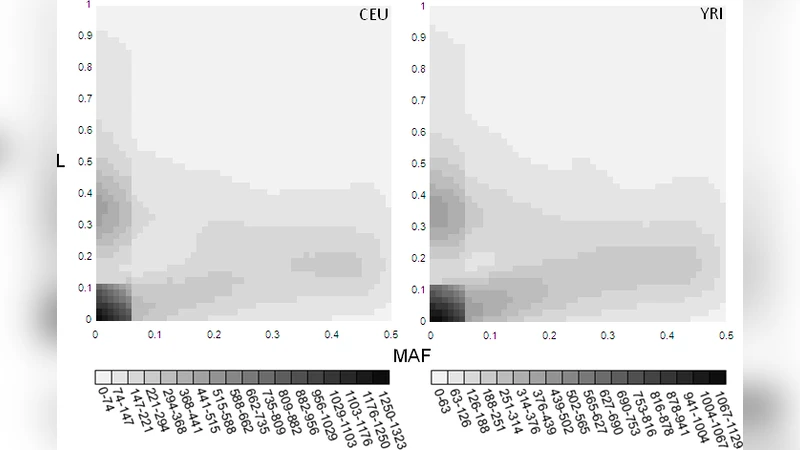
Using the HapMap Phase II SNP catalogue (≈4.07 million SNPs) and the human reference genome (hg18), the authors scanned every annotated donor and acceptor splice site with information‑weight matrices derived from known splice motifs. The average information content of natural sites was 9.8118 bits for donors and 8.1214 bits for acceptors. For each SNP they computed ΔRi at any splice site it intersected, classified the effect (loss of natural site, gain of cryptic site, or both), and retained only changes ≥0.1 bit as potentially functional. They then combined ΔRi with the SNP’s minor‑allele frequency (MAF) to calculate L for each variant.
The analysis identified 1,093,474 SNPs that altered splice‑site information, of which 9,051 affected natural donor or acceptor sites in 5,970 genes. Approximately 70 % of the variants produced ΔRi ≥ 0.5 bits; many of these carried high genetic loads (L > 0.5) despite being relatively common (MAF > 0.05). The authors observed that rare alleles (MAF ≤ 0.05) spanned the full range of loads (0–0.98), whereas common alleles were constrained to lower loads, suggesting purifying selection against strongly deleterious splicing changes. Conversely, some common variants displayed high loads, implying that selection may tolerate moderate splicing perturbations or that compensatory mechanisms exist.
To validate the predictions, the authors integrated expression data from GEO (GSE 7792) and Affymetrix exon arrays for HapMap cell lines. They calculated a splicing index (SI) for each exon (probe‑set intensity normalized by overall gene intensity) and compared SI changes across genotypes with the predicted ΔRi. While SNPs with ΔRi < 0.5 bits rarely produced detectable SI differences, those with ΔRi between 0.7 and 0.9 bits showed a modest but statistically significant trend toward reduced SI in the allele predicted to weaken the splice site. However, the correlation was not strong, reflecting the limited sensitivity of bulk expression assays to subtle splicing alterations and the influence of other regulatory factors.
The paper discusses several limitations. First, the model treats each splice site independently, ignoring linkage disequilibrium and epistatic interactions that could amplify or buffer the effect of multiple nearby SNPs. Second, the fitness‑information relationship is simplified to a logarithmic conversion, which may not capture the full complexity of cellular replication dynamics. Third, only canonical donor/acceptor motifs are considered; auxiliary splicing enhancers, silencers, and RNA‑binding protein sites are omitted, potentially underestimating the functional impact of many variants. Despite these caveats, the study demonstrates that information‑theoretic metrics can be combined with population allele frequencies to generate a biologically meaningful measure of genetic load for splicing‑related SNPs.
In conclusion, the authors provide a scalable, genome‑wide method for prioritizing SNPs that are likely to affect splicing and thus contribute to disease risk or evolutionary fitness. Their approach bridges molecular predictions with population genetics, offering a framework that could be extended to other regulatory elements (e.g., transcription factor binding sites) and refined with more comprehensive models of gene regulation and epistasis.
Comments & Academic Discussion
Loading comments...
Leave a Comment