Azobenzene at Coinage Metal Surfaces: The Role of Dispersive van der Waals Interactions
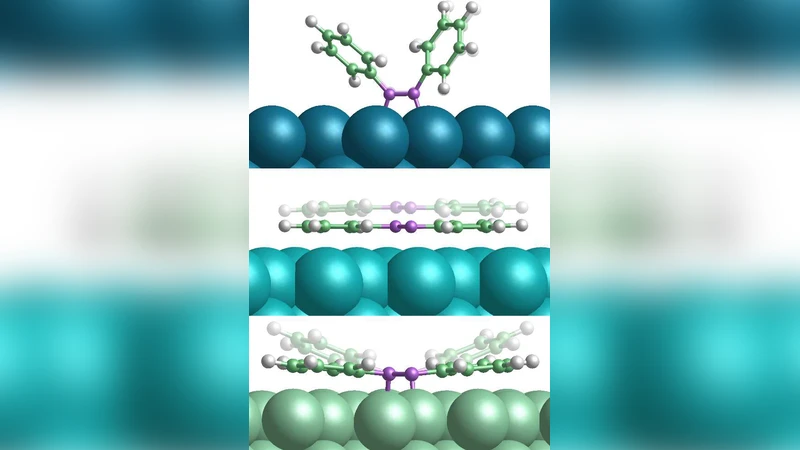
We use different semi-empirical dispersion correction schemes to assess the role of long-range van der Waals interactions in the adsorption of the prototypical molecular switch azobenzene (C6H5-N2-C6H5) at the coinage metal surfaces Cu(111), Ag(111) and Au(111). Compared to preceding density-functional theory results employing a semi-local exchange and correlation functional we obtain partly sizable changes of the computed adsorption geometry and energetics. The discomforting scatter in the results provided by the different schemes is largely attributed to the unknown form of the damping function in the semi-empirical correction expression. Using the congeneric problem of the adsorption of benzene as a vehicle to connection with experiment, we cautiously conclude that the account of dispersive interactions at the metal surfaces provided by the various schemes is in the right ballpark, with the more recent, general schemes likely to overbind.
💡 Research Summary
This paper investigates how long‑range van der Waals (vdW) dispersion forces influence the adsorption of the prototypical molecular switch azobenzene on the (111) facets of the coinage metals Cu, Ag, and Au. The authors apply four semi‑empirical dispersion‑correction schemes—DFT‑D2, DFT‑D3, DFT‑D3(BJ), and the Tkatchenko‑Scheffler (TS) method—to density‑functional theory calculations that otherwise use a semi‑local exchange‑correlation functional (PBE). By comparing the results from each scheme with the plain PBE outcomes, they quantify changes in adsorption geometry (height above the surface, tilt of the phenyl rings, N=N bond length) and binding energy.
Inclusion of dispersion consistently reduces the molecule‑surface distance: Cu(111) shows a decrease of roughly 0.3 Å, Ag(111) about 0.15 Å, and Au(111) about 0.10 Å. The N=N bond shortens slightly on Cu, indicating a stronger interaction. Binding energies are also enhanced: while PBE predicts ≈‑0.8 eV, D3 and TS raise this to ≈‑1.2 eV and ≈‑1.1 eV respectively, whereas D2 overbinds to ≈‑1.4 eV. The spread among the schemes is traced back to the poorly defined damping function that moderates the short‑range part of the correction; different functional forms lead to appreciable variations.
To benchmark the theoretical predictions, the authors turn to benzene adsorption on the same metal surfaces, for which reliable experimental adsorption energies exist (≈‑0.64 eV for benzene/Au(111)). TS and D3 reproduce the experimental value within 0.1–0.2 eV, while D2 overestimates binding by about 0.2 eV. This comparison suggests that the newer, more general dispersion schemes are in the right ball‑park but tend to overbind slightly.
The study concludes that vdW dispersion forces are essential for an accurate description of azobenzene adsorption, especially on the more reactive Cu and Ag surfaces. However, the current semi‑empirical corrections still suffer from methodological uncertainty, primarily due to the arbitrary choice of damping functions. The authors recommend systematic benchmarking against experimental data and the development of non‑empirical approaches—such as the random phase approximation (RPA) or many‑body dispersion (MBD)—that treat electronic screening and dispersion on an equal footing. Such advances will be crucial for reliable modeling of metal‑organic interfaces in molecular electronics and surface‑mounted switch devices.
Comments & Academic Discussion
Loading comments...
Leave a Comment