A Molecular Study of CaCO$_3$ cluster configurations
Equilibrium relationships involving solids are based on bulk thermodynamic properties that concern ideal crystals of infinite size. However, real processes towards equilibrium imply development of finite molecular-scale entities. The configuration of these early-stage clusters and the estimation of their excess energies with respect to the ideal crystal are keys to understanding the macroscopic behaviour of a given system. As nucleation events are difficult to study experimentally, both because they occur spontaneously and because the nucleus size is very small, atomistic simulations are a suitable tool for understanding the early stages of crystallisation. Here, starting from the ideal atomic positions in calcite and aragonite, the relaxation in vacuum of finite clusters of CaCO$_3$ is explored. Nucleation and growth of calcium carbonate phases constitute a very important subject of research in a wide variety of fields. A complete study of CaCO$_3$ should include many different aspects: size and shape of the critical nuclei under diverse conditions, possibility of nucleation from precursor phases, nucleus energy and nucleus surface energy, relationship nucleus-substrate in heterogeneous nucleation…We present a preliminary study of nucleation of calcium carbonate where nuclei are considered to be isolated from any previous phase or substrate. Even when this situation does in no way represent realistic conditions, it can be a helpful first approach to more complex studies.
💡 Research Summary
This paper investigates the atomistic energetics and structural evolution of calcium carbonate (CaCO₃) clusters ranging from a single formula unit up to 2 000 units, using vacuum‑isolated models. Starting from the ideal bulk positions of the three polymorphs—calcite, aragonite, and vaterite—the authors generated spherical, charge‑neutral clusters and performed full geometry optimizations at constant pressure with the GULP code. The short‑range interactions were described by a comprehensive force field derived by Rohl et al., which combines Buckingham, Morse, harmonic spring, three‑body, and out‑of‑plane terms, while long‑range electrostatics were treated with a shell model for oxygen to capture polarizability. To validate the force field for very small aggregates, clusters containing ten or fewer formula units were also optimized with the DFT‑based SIESTA package; the resulting energies and atomic coordinates agreed closely with the GULP results.
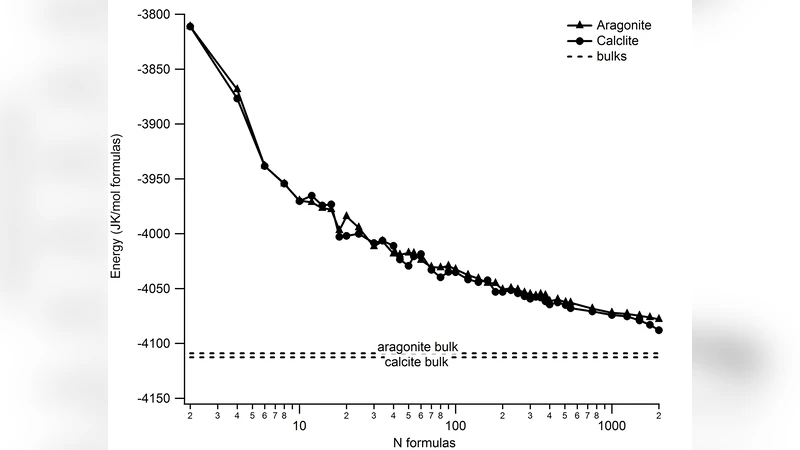
The study distinguishes between “unrelaxed” clusters (the initial bulk‑derived geometry) and “relaxed” clusters (after full energy minimization). For unrelaxed clusters, the energy per formula unit drops sharply as the cluster grows, but the trend is punctuated by size‑specific stability peaks—for example, a calcite cluster of 24 units is more stable than those of 30 or 34 units, and an aragonite cluster of 10 units is more stable than those of 12, 14, or 16 units. These anomalies are most pronounced for clusters containing fewer than about 30 formula units, reflecting the disproportionate influence of surface atoms and incomplete Ca coordination polyhedra.
Relaxation reduces the overall energy and smooths the size‑dependence, yet even the largest simulated cluster (2 000 units) remains about 25 kJ mol⁻¹ (calcite‑derived) or 30 kJ mol⁻¹ (aragonite‑derived) higher than the corresponding bulk lattice energy calculated with the same force field. This residual excess indicates that surface effects persist even for clusters approaching nanometer dimensions.
Based on the energetic and structural data, the authors categorize the clusters into three size regimes:
-
Small clusters (≤12 formula units). All atoms lie at the surface, Ca coordination is incomplete, and the relaxed structures are essentially amorphous. Simulated X‑ray diffraction (XRD) patterns show no discernible Bragg peaks, confirming the lack of long‑range order.
-
Intermediate clusters (14–160 formula units). The interior begins to retain the alternating Ca–CO₃ layering characteristic of the parent polymorph, while the surface undergoes substantial rearrangement. CO₃ groups tend to lie tangent to the surface, and surface Ca atoms acquire additional O contacts. Depending on the initial polymorph, either calcite‑derived or aragonite‑derived clusters can be energetically favored; notably, some aragonite‑derived clusters are more stable than their calcite counterparts in this size range. Simulated XRD patterns display weakened but recognizable reflections of the original bulk phase.
-
Large clusters (≥170 formula units). The core structure closely matches the bulk crystal, with only surface atoms deviating. All clusters derived from a calcite starting geometry are slightly lower in energy than those derived from aragonite. The XRD patterns of these large clusters are virtually indistinguishable from those of the corresponding bulk polymorphs, indicating that experimental diffraction could readily identify the underlying phase.
The authors discuss the implications for nucleation theory. The finding that sub‑12‑unit clusters are amorphous supports the experimentally observed existence of amorphous calcium carbonate (ACC) as a precursor to crystalline phases. Moreover, the occasional energetic advantage of aragonite‑derived clusters in the intermediate size regime provides a plausible atomistic rationale for the frequent observation of aragonite or other metastable polymorphs during the early stages of calcite crystallization, contrary to the simple Ostwald stepwise transformation (ACC → vaterite → aragonite → calcite). The persistent surface‑related energy penalty, even for large clusters, highlights the limitation of classical nucleation theory, which assumes a size‑independent surface tension and a perfectly spherical nucleus.
In conclusion, the paper demonstrates that (i) very small CaCO₃ nuclei are intrinsically amorphous, (ii) intermediate nuclei can retain polymorph‑specific ordering but are strongly influenced by surface relaxation, and (iii) large nuclei approach bulk energetics but never fully reach them due to residual surface effects. These results underscore the necessity of atomistic simulations for a realistic description of nucleation pathways in calcium carbonate systems and lay the groundwork for future studies that incorporate solvent, ion‑pairing, and heterogeneous substrate effects.
Comments & Academic Discussion
Loading comments...
Leave a Comment