Towards a theoretical understanding of false positives in DNA motif finding
Detection of false-positive motifs is one of the main causes of low performance in motif finding methods. It is generally assumed that false-positives are mostly due to algorithmic weakness of motif-finders. Here, however, we derive the theoretical dependence of false positives on dataset size and find that false positives can arise as a result of large dataset size, irrespective of the algorithm used. Interestingly, the false-positive strength depends more on the number of sequences in the dataset than it does on the sequence length. As expected, false-positives can be reduced by decreasing the sequence length or by adding more sequences to the dataset. The dependence on number of sequences, however, diminishes and reaches a plateau after which adding more sequences to the dataset does not reduce the false-positive rate significantly. Based on the theoretical results presented here, we provide a number of intuitive rules of thumb that may be used to enhance motif-finding results in practice.
💡 Research Summary
The paper addresses a pervasive problem in DNA motif discovery: the appearance of strong‑looking motifs that are in fact false positives. While many studies attribute this to weaknesses in motif‑finding algorithms, the authors demonstrate that the statistical properties of large data sets alone can generate such spurious motifs, regardless of the algorithm used.
Using the “one‑occurrence‑per‑sequence” (OOPS) model, they treat each of the n sequences (length L) as being generated from a background nucleotide distribution g (e.g., uniform or genome‑wide frequencies). A candidate motif is represented by a probability matrix f of width W, and its strength is measured by the Kullback‑Leibler divergence D(f,g), also called information content. By applying large‑deviation theory, specifically Sanov’s theorem, they derive an analytical expression for the expected sequence length at which a motif of strength D is likely to appear by chance:
L ≈ (W² · D · (n + 1) · W · (|A| − 1)) / n (Equation 2)
Here |A| is the alphabet size (4 for DNA). The equation predicts that when the actual sequence length exceeds this L, a motif with information content D will almost surely be observed even in completely random data.
Key implications of the formula:
-
Number of sequences (n) matters more than sequence length (L). For a fixed L, tripling n reduces the detectable false‑positive strength as much as increasing L by two orders of magnitude. However, the benefit of adding more sequences plateaus: beyond roughly 30–50 sequences the reduction in D becomes marginal.
-
Motif width (W) scales linearly with information content. For a given D, shorter motifs are easier to detect and less prone to false positives.
-
Sequence length still matters, but its effect is weaker than that of n. Shortening L modestly lowers the expected false‑positive strength, but the dominant lever is the number of independent sequences.
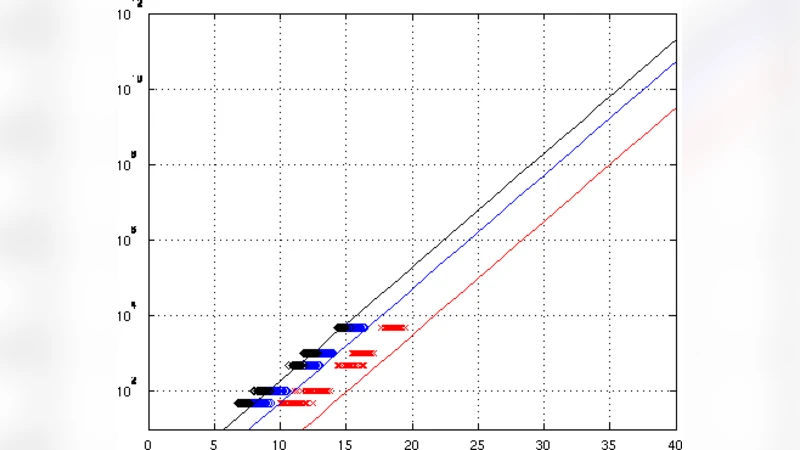
To validate the theory, the authors ran extensive simulations with the MEME software (configured in OOPS mode). They generated random DNA sets for various combinations of n (10, 20, 30), W (5, 10, 15), and L (50–5000). For each condition they performed 50 Monte‑Carlo runs, extracted the most significant motif reported by MEME, and computed its D. The empirical D values (plotted as stars) aligned closely with the theoretical curves (solid lines) in Figure 2, confirming the model’s accuracy.
From these results they distilled five practical “rules of thumb” for experimental design:
- Prefer shorter sequences when biologically feasible to limit the search space.
- Increase the number of sequences; moving from 10 to 30 sequences can cut the expected false‑positive strength by >6 bits for W = 10, but gains diminish after ~30–50 sequences.
- Prioritize adding sequences over extending sequence length, because n has a stronger impact on false‑positive reduction.
- Use Equation 2 (or the curves in Fig. 2) to estimate the expected false‑positive strength for a given dataset; motifs that do not substantially exceed this threshold should be treated with suspicion.
- Target narrower motifs when possible, as they are less likely to be mimicked by random background.
The authors also discuss extensions. Replacing |A| = 4 with |A| = 20 (protein alphabet) makes L grow exponentially, implying that false‑positive rates in protein motif discovery are intrinsically lower under comparable settings. They outline how the derivation can be adapted to other motif‑finding models such as ZOOPS (zero‑or‑one occurrence per sequence).
Finally, as a side product they provide a simple conservative p‑value approximation:
p ≈ (n + 1)·W·(|A| − 1)·2^(−n·D) (Equation 3)
This formula offers a quick way to assess statistical significance without resorting to computationally intensive exact calculations.
In summary, the paper presents a clear theoretical framework that links dataset size, motif width, and background entropy to the likelihood of observing strong false‑positive motifs. By confirming the theory with MEME simulations and translating the results into actionable guidelines, the work offers both a deeper understanding of the statistical limits of motif discovery and practical advice for designing more reliable experiments.
Comments & Academic Discussion
Loading comments...
Leave a Comment