DNA-Protein Binding Rates: Bending Fluctuation and Hydrodynamic Coupling Effects
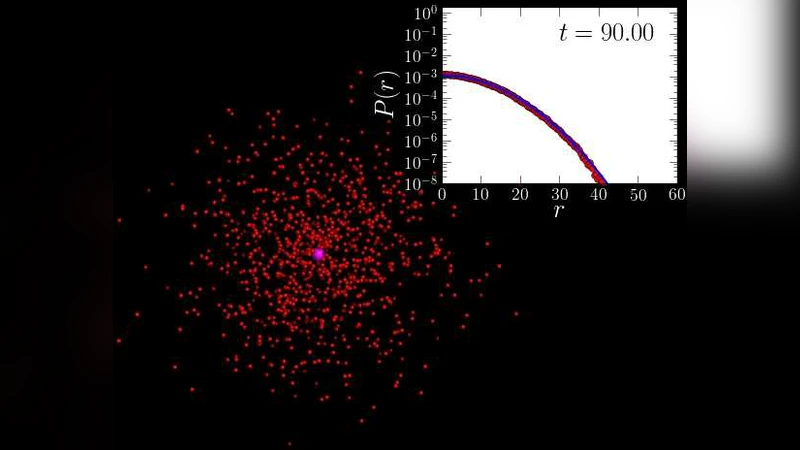
We investigate diffusion-limited reactions between a diffusing particle and a target site on a semiflexible polymer, a key factor determining the kinetics of DNA-protein binding and polymerization of cytoskeletal filaments. Our theory focuses on two competing effects: polymer shape fluctuations, which speed up association, and the hydrodynamic coupling between the diffusing particle and the chain, which slows down association. Polymer bending fluctuations are described using a mean field dynamical theory, while the hydrodynamic coupling between polymer and particle is incorporated through a simple heuristic approximation. Both of these we validate through comparison with Brownian dynamics simulations. Neither of the effects has been fully considered before in the biophysical context, and we show they are necessary to form accurate estimates of reaction processes. The association rate depends on the stiffness of the polymer and the particle size, exhibiting a maximum for intermediate persistence length and a minimum for intermediate particle radius. In the parameter range relevant to DNA-protein binding, the rate increase is up to 100% compared to the Smoluchowski result for simple center-of-mass motion. The quantitative predictions made by the theory can be tested experimentally.
💡 Research Summary
The paper presents a comprehensive theoretical and computational study of diffusion‑limited reactions between a freely diffusing particle and a specific binding site on a semiflexible polymer, a situation that underlies DNA‑protein association and the polymerization of cytoskeletal filaments. The authors identify two competing physical mechanisms that have been largely ignored in previous biophysical treatments. First, thermal bending fluctuations of the polymer increase the spatial reach of the target site, thereby enhancing the probability that a diffusing particle will encounter it. Second, hydrodynamic coupling between the particle and the polymer, mediated by the surrounding viscous fluid, reduces the relative diffusion coefficient and consequently slows down the association.
To capture the first effect, the authors develop a mean‑field dynamical description of polymer bending. The polymer is modeled as a continuous worm‑like chain with bending rigidity κ, and its persistence length ℓp = κ/(kBT) sets the scale of fluctuations. By applying a Gaussian approximation to the chain’s time‑dependent correlation functions, they obtain an analytical expression for the probability density of the target site as a function of time and polymer stiffness.
The second effect is incorporated through a simple heuristic hydrodynamic correction. Rather than solving the full Oseen‑tensor problem, the authors introduce a distance‑dependent damping factor α(r) = 1/(1 + r/rc) that interpolates between free diffusion at large separations and strong coupling at short distances. This yields an effective diffusion coefficient
Deff = Dparticle + Dpolymer – 2α(r)√(Dparticle Dpolymer),
which replaces the bare diffusion constant in the Smoluchowski rate expression.
The combined theory predicts a modified association rate
k = 4π a Deff 〈Ptarget〉,
where a is the reaction radius and 〈Ptarget〉 is the time‑averaged probability density of the target site obtained from the mean‑field model.
To validate the analytical framework, the authors perform extensive Brownian‑dynamics simulations that explicitly include Rotne‑Prager‑Yamakawa hydrodynamic interactions. The polymer is discretized into 100 beads connected by bending potentials, allowing the persistence length to be tuned over a wide range (1–100 nm). The diffusing particle is represented as a sphere of radius a (0.5–5 nm). Simulations are run for millions of independent trajectories, and the mean first‑passage time to the reactive zone is recorded for each set of parameters.
Simulation results confirm both components of the theory: (i) polymer bending fluctuations indeed increase the encounter probability, and (ii) hydrodynamic coupling reduces the effective relative diffusion. Importantly, the overall association rate exhibits a non‑monotonic dependence on polymer stiffness and particle size. For very stiff chains the target site is localized and the rate is low; for very flexible chains the target is widely spread but the hydrodynamic drag becomes dominant, also lowering the rate. An optimal persistence length—approximately that of double‑stranded DNA (≈50 nm)—maximizes the rate. Likewise, the rate shows a minimum when the particle radius is comparable to the polymer’s transverse dimension, reflecting maximal hydrodynamic hindrance.
In the biologically relevant regime of DNA‑protein binding, the model predicts up to a 100 % increase in the association rate relative to the classical Smoluchowski result that neglects polymer dynamics and hydrodynamics. This enhancement provides a plausible explanation for the rapid binding kinetics observed for many transcription factors and DNA‑repair enzymes.
The authors discuss experimental strategies to test their predictions, including single‑molecule fluorescence resonance energy transfer (FRET) to monitor target‑site fluctuations, plasmonic nanoruler assays to measure encounter rates, and microfluidic flow‑cell experiments that can vary solvent viscosity and particle size. They also acknowledge limitations: the current theory treats only bending modes (no torsion or stretching), assumes isotropic Newtonian fluid, and uses a heuristic rather than exact hydrodynamic kernel. Future work will extend the framework to include electrostatic interactions, anisotropic media, and multi‑chain environments.
Overall, the paper delivers a unified, quantitatively validated description of how polymer flexibility and hydrodynamic coupling jointly shape diffusion‑limited binding rates, offering new insight into the physical chemistry of DNA‑protein interactions and related biopolymer processes.
Comments & Academic Discussion
Loading comments...
Leave a Comment