Boolean Threshold Networks: Virtues and Limitations for Biological Modeling
Boolean threshold networks have recently been proposed as useful tools to model the dynamics of genetic regulatory networks, and have been successfully applied to describe the cell cycles of \textit{S. cerevisiae} and \textit{S. pombe}. Threshold networks assume that gene regulation processes are additive. This, however, contrasts with the mechanism proposed by S. Kauffman in which each of the logic functions must be carefully constructed to accurately take into account the combinatorial nature of gene regulation. While Kauffman Boolean networks have been extensively studied and proved to have the necessary properties required for modeling the fundamental characteristics of genetic regulatory networks, not much is known about the essential properties of threshold networks. Here we study the dynamical properties of these networks with different connectivities, activator-repressor proportions, activator-repressor strengths and different thresholds. Special attention is paid to the way in which the threshold value affects the dynamical regime in which the network operates and the structure of the attractor landscape. We find that only for a very restricted set of parameters, these networks show dynamical properties consistent with what is observed in biological systems. The virtues of these properties and the possible problems related with the restrictions are discussed and related to earlier work that uses these kind of models.
💡 Research Summary
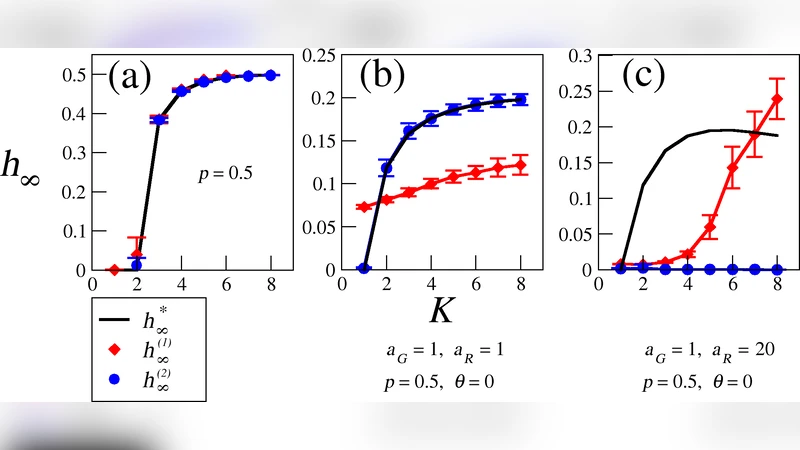
The paper provides a systematic investigation of Boolean threshold networks (TN) as an alternative to the classic Kauffman Boolean networks for modeling genetic regulatory systems. While Kauffman’s approach assigns a specific logical function to each gene, TN assumes that the regulatory effect of all inputs is additive: the weighted sum of incoming signals is compared to a preset threshold, and the gene’s state is set to 0 or 1 accordingly. The authors explore how four key parameters—average connectivity (K), the proportion of activators versus repressors (p), the relative strengths of activators (α) and repressors (β), and the threshold value (θ)—shape the dynamical regime of the network.
Using the average sensitivity λ as the order parameter, they map out ordered (λ < 1), critical (λ ≈ 1), and chaotic (λ > 1) regions. Their simulations show that only a narrow band of parameter combinations yields dynamics compatible with biological observations. Specifically, low to moderate connectivity (K = 2–4), a balanced activator‑repressor ratio (p ≈ 0.5), comparable strengths (α ≈ β), and a threshold set to roughly 40‑60 % of the mean input sum keep λ near unity. In this regime the attractor landscape consists of a small number of short‑period cycles (≤10 steps) and exhibits robustness to perturbations, mirroring the limited number of cell‑cycle states seen in yeast.
When θ is too low, most nodes stay “on,” λ exceeds one, and the system falls into a chaotic regime with a profusion of long‑period attractors. Conversely, an excessively high θ forces most nodes “off,” driving the network into an ordered regime where almost all trajectories collapse to a single fixed point. The model is also highly sensitive to the activator‑repressor proportion: strong imbalance (p < 0.2 or p > 0.8) pushes the system away from criticality, either freezing dynamics or amplifying noise. Moreover, increasing K beyond four or introducing large asymmetries between α and β rapidly expands the chaotic region, making the network’s behavior biologically implausible.
The authors discuss the virtues of TN: its mathematical simplicity, ease of parameterization, and the fact that many regulatory interactions can be approximated as additive. These features have enabled successful applications to the cell‑cycle networks of S. cerevisiae and S. pombe. However, the study also highlights serious limitations. The additive assumption cannot capture the combinatorial logic (AND, OR, NOT) that is intrinsic to many transcriptional modules, leading to an oversimplified representation of gene regulation. The dynamical behavior is confined to a tight parameter space, requiring fine‑tuned thresholds and balanced activator‑repressor ratios—conditions that may not hold across diverse biological contexts. High connectivity or strong strength asymmetries, common in real regulatory networks, drive the model into unrealistic chaotic or frozen regimes.
In conclusion, Boolean threshold networks can reproduce key qualitative features of genetic regulatory dynamics, but only under restrictive conditions. The paper suggests future extensions such as incorporating nonlinear activation functions, multiple thresholds per node, or hybrid schemes that blend additive and logical components. Such refinements could broaden the applicability of TN while preserving its computational tractability, making it a more robust tool for systems‑biology modeling.
Comments & Academic Discussion
Loading comments...
Leave a Comment