Using GWAS Data to Identify Copy Number Variants Contributing to Common Complex Diseases
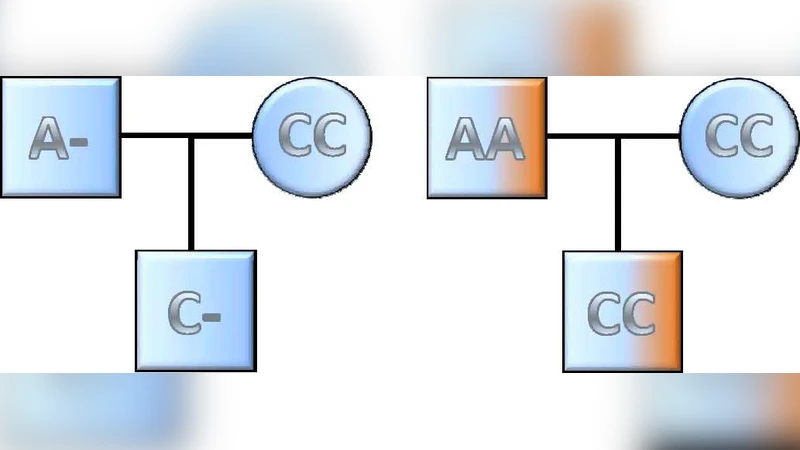
Copy number variants (CNVs) account for more polymorphic base pairs in the human genome than do single nucleotide polymorphisms (SNPs). CNVs encompass genes as well as noncoding DNA, making these polymorphisms good candidates for functional variation. Consequently, most modern genome-wide association studies test CNVs along with SNPs, after inferring copy number status from the data generated by high-throughput genotyping platforms. Here we give an overview of CNV genomics in humans, highlighting patterns that inform methods for identifying CNVs. We describe how genotyping signals are used to identify CNVs and provide an overview of existing statistical models and methods used to infer location and carrier status from such data, especially the most commonly used methods exploring hybridization intensity. We compare the power of such methods with the alternative method of using tag SNPs to identify CNV carriers. As such methods are only powerful when applied to common CNVs, we describe two alternative approaches that can be informative for identifying rare CNVs contributing to disease risk. We focus particularly on methods identifying de novo CNVs and show that such methods can be more powerful than case-control designs. Finally we present some recommendations for identifying CNVs contributing to common complex disorders.
💡 Research Summary
The paper provides a comprehensive review of how copy‑number variants (CNVs) can be identified and leveraged in genome‑wide association studies (GWAS) to uncover genetic contributors to common complex diseases. It begins by emphasizing that CNVs encompass more polymorphic base pairs in the human genome than single‑nucleotide polymorphisms (SNPs) and often involve entire genes or regulatory regions, making them strong candidates for functional variation.
The authors then describe the two primary signals generated by high‑throughput genotyping platforms: the log‑R ratio (LRR), which reflects hybridization intensity and therefore total copy number, and the B‑allele frequency (BAF), which captures allelic imbalance. By jointly modeling LRR and BAF, researchers can infer discrete copy‑number states (0, 1, 2, 3, …) across the genome.
A substantial portion of the review is devoted to statistical methods for CNV detection. Classical approaches such as Hidden Markov Models (HMM) treat the genome as a sequence of hidden copy‑number states and use emission probabilities derived from LRR/BAF to segment the data. Gaussian Mixture Models (GMM) assume that each copy‑number class follows a normal distribution and employ Expectation‑Maximization to estimate parameters. More recent developments include Bayesian hierarchical models that incorporate prior information about CNV frequency and length, as well as deep‑learning frameworks that learn complex, non‑linear patterns from raw intensity data. The authors compare these methods in terms of sensitivity, specificity, computational cost, and robustness to noise, concluding that hybrid intensity‑based approaches generally outperform tag‑SNP strategies for common CNVs.
For common CNVs (minor allele frequency >5 %), the paper discusses the “tag‑SNP” approach, wherein a SNP in strong linkage disequilibrium with a CNV serves as a surrogate marker. While cost‑effective, this method cannot distinguish deletions from duplications and may miss CNVs that are not well tagged by surrounding SNPs. Direct intensity‑based detection therefore remains the preferred strategy when high‑quality LRR/BAF data are available.
Rare CNVs (MAF <1 %) pose a different challenge because case‑control designs lack sufficient power. The authors advocate for de‑novo CNV discovery using parent‑offspring trios, which isolates newly arisen variants that are enriched in affected individuals. They also highlight the integration of next‑generation sequencing read‑depth, split‑read, and discordant‑pair signals to capture small or complex CNVs that microarray platforms may overlook.
Quality control (QC) procedures are presented in detail. The review stresses the importance of background correction, probe filtering, and the use of QC metrics such as LRR standard deviation, BAF drift, and waviness factor. Multiple testing correction strategies—including false discovery rate (FDR), Bonferroni, and permutation‑based methods—are recommended to control type‑I error.
Finally, the authors provide practical recommendations: (1) combine several detection algorithms and generate a consensus call set; (2) validate findings in independent cohorts; (3) deposit CNV calls in public repositories to facilitate meta‑analysis; and (4) explore integrative analyses that couple CNV data with transcriptomics, epigenomics, and proteomics to elucidate functional consequences. They also suggest that future work should focus on building standardized pipelines, expanding reference CNV databases, and applying machine‑learning models for personalized risk prediction.
In summary, the paper serves as a roadmap for researchers aiming to harness GWAS data for CNV discovery, offering methodological guidance, comparative performance assessments, and strategic advice for both common and rare variant analyses in the context of complex disease genetics.
Comments & Academic Discussion
Loading comments...
Leave a Comment