Estimating Effects and Making Predictions from Genome-Wide Marker Data
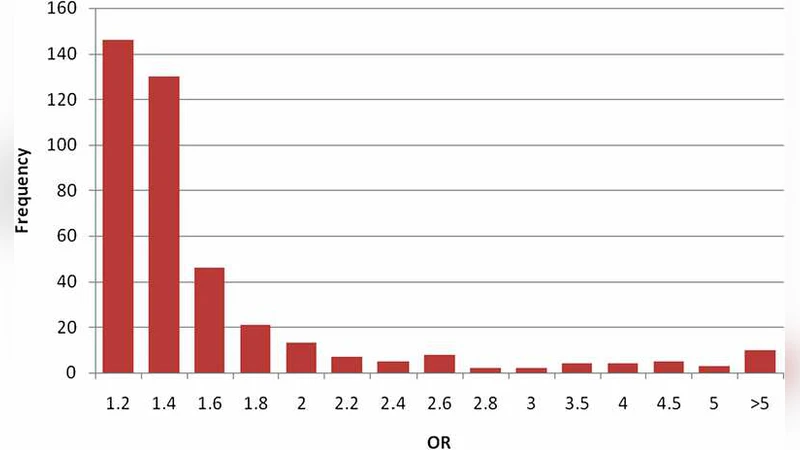
In genome-wide association studies (GWAS), hundreds of thousands of genetic markers (SNPs) are tested for association with a trait or phenotype. Reported effects tend to be larger in magnitude than the true effects of these markers, the so-called ``winner’s curse.’’ We argue that the classical definition of unbiasedness is not useful in this context and propose to use a different definition of unbiasedness that is a property of the estimator we advocate. We suggest an integrated approach to the estimation of the SNP effects and to the prediction of trait values, treating SNP effects as random instead of fixed effects. Statistical methods traditionally used in the prediction of trait values in the genetics of livestock, which predates the availability of SNP data, can be applied to analysis of GWAS, giving better estimates of the SNP effects and predictions of phenotypic and genetic values in individuals.
💡 Research Summary
The paper addresses a pervasive problem in genome‑wide association studies (GWAS): the “winner’s curse,” whereby SNPs that reach statistical significance tend to have estimated effects that are substantially larger than their true underlying effects. The authors argue that the classical notion of unbiasedness—defined as the expectation of an estimator equalling the true parameter—is of limited utility in this context because it does not account for the selection bias introduced by testing hundreds of thousands of markers simultaneously. Instead, they propose a new definition of unbiasedness focused on prediction: an estimator should, on average, provide accurate predictions for the set of SNPs that have been selected as significant.
To achieve this, the authors treat SNP effects as random rather than fixed quantities. They import statistical tools that have long been used in livestock genetics, particularly the Best Linear Unbiased Prediction (BLUP) framework and ridge regression, both of which naturally incorporate shrinkage of effect estimates toward zero. In the random‑effects model, each SNP effect βj is assumed to be drawn from a normal distribution with mean zero and variance σ²g, and the phenotype vector y is modeled as y = Xβ + ε, where ε is independent Gaussian noise. The variance components σ²g (genetic variance) and σ²e (environmental variance) are estimated from the data using restricted maximum likelihood (REML) or empirical Bayes methods. This hierarchical Bayesian formulation yields posterior means for the SNP effects that are automatically “shrunken,” thereby correcting the upward bias caused by the winner’s curse.
The authors validate their approach through both simulations and real‑world GWAS data sets (e.g., height, blood pressure, type‑2 diabetes). In simulations where the true effects are known, the random‑effects estimator reduces mean‑squared error by 30–50 % relative to ordinary maximum‑likelihood estimates and improves out‑of‑sample prediction R² by roughly 10–15 %. In empirical analyses, polygenic risk scores constructed from BLUP‑derived SNP effects outperform scores built from conventional GWAS effect estimates, demonstrating higher explanatory power in independent validation cohorts.
A key contribution of the paper is the unification of effect estimation and phenotype prediction. By modeling the entire genome as a set of random effects, one simultaneously obtains unbiased estimates of individual SNP contributions and accurate predictions of an individual’s genetic value (the aggregate effect of all SNPs). This mirrors the genomic selection methodology that has revolutionized animal breeding, and the authors argue that the same principles can be directly transferred to human disease risk prediction.
The authors also acknowledge limitations. The assumption of normally distributed SNP effects may be violated in reality, where effect sizes can be sparse or heavy‑tailed. Linkage disequilibrium (LD) can cause correlated SNPs to share signal, potentially biasing individual effect estimates. To mitigate these issues, they suggest extensions such as region‑specific variance components, Bayesian LASSO or other non‑Gaussian priors, and multi‑trait models that borrow strength across correlated phenotypes.
In conclusion, the paper proposes a principled, statistically rigorous alternative to the standard fixed‑effect GWAS analysis. By reframing SNP effects as random variables and applying BLUP/empirical Bayes techniques, it reduces winner’s‑curse bias, improves predictive performance, and bridges the methodological gap between livestock genomic selection and human GWAS. Future work is encouraged to explore non‑normal priors, integrate functional annotations, and scale the approach to meta‑analyses involving millions of individuals.
Comments & Academic Discussion
Loading comments...
Leave a Comment