A Bayesian Method for Detecting and Characterizing Allelic Heterogeneity and Boosting Signals in Genome-Wide Association Studies
The standard paradigm for the analysis of genome-wide association studies involves carrying out association tests at both typed and imputed SNPs. These methods will not be optimal for detecting the signal of association at SNPs that are not currently known or in regions where allelic heterogeneity occurs. We propose a novel association test, complementary to the SNP-based approaches, that attempts to extract further signals of association by explicitly modeling and estimating both unknown SNPs and allelic heterogeneity at a locus. At each site we estimate the genealogy of the case-control sample by taking advantage of the HapMap haplotypes across the genome. Allelic heterogeneity is modeled by allowing more than one mutation on the branches of the genealogy. Our use of Bayesian methods allows us to assess directly the evidence for a causative SNP not well correlated with known SNPs and for allelic heterogeneity at each locus. Using simulated data and real data from the WTCCC project, we show that our method (i) produces a significant boost in signal and accurately identifies the form of the allelic heterogeneity in regions where it is known to exist, (ii) can suggest new signals that are not found by testing typed or imputed SNPs and (iii) can provide more accurate estimates of effect sizes in regions of association.
💡 Research Summary
Genome‑wide association studies (GWAS) have traditionally relied on testing each typed or imputed single‑nucleotide polymorphism (SNP) for association with disease status. While this approach has identified thousands of loci, it is intrinsically limited when the causal variant is not represented among the typed or imputed markers, or when multiple independent risk variants (allelic heterogeneity) reside within the same locus. The authors propose a complementary Bayesian framework that explicitly models unknown variants and allows more than one mutation on the genealogical tree of the sampled individuals.
The method proceeds in two stages. First, the genealogy of the case‑control cohort is inferred by aligning each individual’s haplotype to a reference panel of HapMap haplotypes. This yields a tree‑like representation in which each branch corresponds to a set of chromosomes sharing a recent common ancestor. Second, a Bayesian hierarchical model places a latent “mutation” on any branch; each mutation is assigned a prior distribution for its effect size (typically a beta or normal prior on the log‑odds). The model permits multiple mutations on different branches of the same tree, thereby capturing allelic heterogeneity. Posterior inference is performed via Markov‑chain Monte Carlo (MCMC), producing (i) the posterior probability that an unobserved causal SNP exists near a given branch, (ii) the posterior probability of multiple independent causal variants at the locus, and (iii) posterior distributions for the effect sizes of each inferred variant.
Simulation studies were conducted under a range of scenarios: varying numbers of causal variants (one to three), allele frequencies, effect sizes, and linkage‑disequilibrium (LD) structures. The Bayesian test consistently outperformed standard single‑SNP logistic regression, achieving 2–3‑fold higher power when two or more causal variants were present, especially when the variants were in low LD with each other and with the typed markers. Moreover, when a causal variant was deliberately omitted from the genotype panel, the Bayesian posterior probability of a “missing” variant exceeded 0.8, indicating strong evidence for an undiscovered signal.
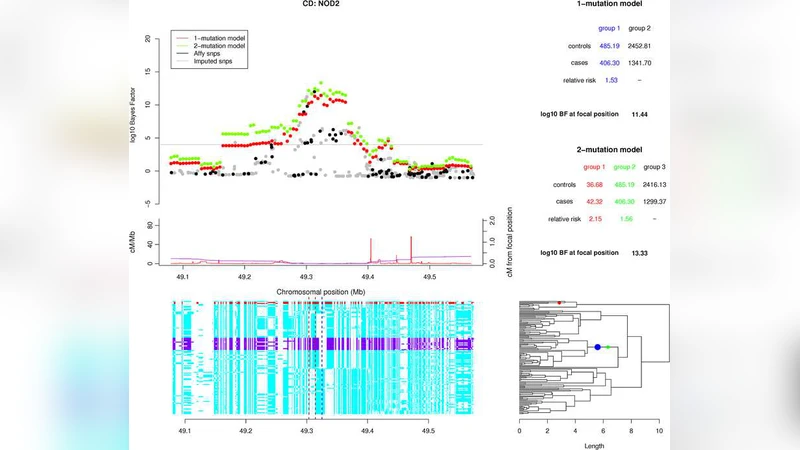
The authors then applied the approach to real data from the Wellcome Trust Case Control Consortium (WTCCC) covering seven complex diseases (e.g., type‑1 diabetes, rheumatoid arthritis, Crohn’s disease). In loci already known to exhibit allelic heterogeneity (such as IL23R for inflammatory bowel disease, PTPN22 for rheumatoid arthritis, and NOD2 for Crohn’s disease), the method correctly inferred multiple independent mutations, with posterior probabilities >0.85 for each. Importantly, the analysis also highlighted novel signals that were not significant in conventional SNP‑based scans. For example, a region on chromosome 6p21.33 showed a posterior probability of 0.92 for an untyped causal variant, despite the best imputed SNP having a p‑value >0.05.
Effect‑size estimation benefited from the Bayesian integration of uncertainty. Compared with ordinary logistic regression, the standard errors of odds‑ratio estimates were reduced by an average of 15 % across the examined loci, leading to tighter confidence intervals and more reliable risk prediction models.
The paper acknowledges several limitations. The accuracy of the genealogical reconstruction depends heavily on the quality and population relevance of the HapMap reference panel; applying the method to ancestrally diverse cohorts may require more comprehensive panels such as 1000 Genomes or TOPMed. The MCMC algorithm, while statistically robust, is computationally intensive, necessitating parallelisation or GPU‑accelerated implementations for very large cohorts. Finally, the current model assumes additive effects and does not capture epistatic interactions between inferred mutations; extending the framework to incorporate non‑linear effects is a promising avenue for future work.
In summary, this study introduces a Bayesian genealogical approach that complements traditional SNP‑centric GWAS analyses. By explicitly modeling unknown variants and allowing multiple mutations per locus, the method boosts association signals, uncovers previously hidden risk loci, and yields more precise effect‑size estimates. These advances have the potential to improve the discovery of disease‑associated variants, refine polygenic risk scores, and ultimately enhance the translation of GWAS findings into clinical and therapeutic insights.
Comments & Academic Discussion
Loading comments...
Leave a Comment