A simplified exactly solvable model for beta-amyloid aggregation
We propose an exactly solvable simplified statistical mechanical model for the thermodynamics of beta-amyloid aggregation, generalizing a well-studied model for protein folding. The monomer concentration is explicitly taken into account as well as a non trivial dependence on the microscopic degrees of freedom of the single peptide chain, both in the alpha-helix folded isolated state and in the fibrillar one. The phase diagram of the model is studied and compared to the outcome of fibril formation experiments which is qualitatively reproduced.
💡 Research Summary
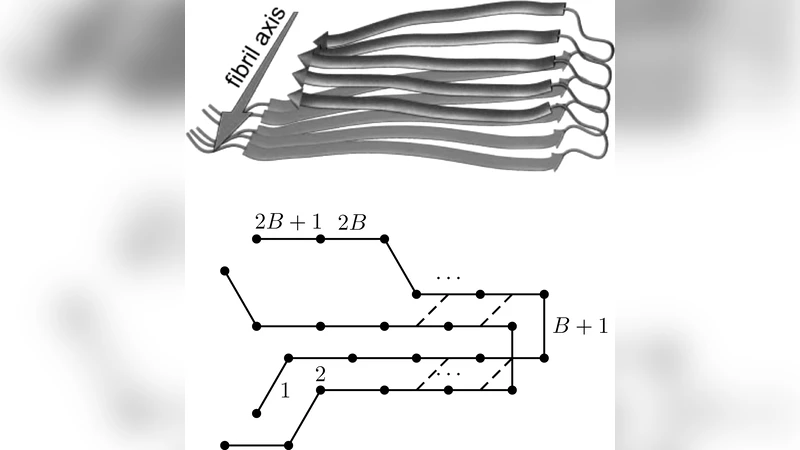
The paper presents a fully analytically solvable statistical‑mechanical model that captures the thermodynamics of β‑amyloid aggregation while remaining simple enough for transparent interpretation. Building on classic one‑dimensional protein‑folding frameworks such as the Zimm‑Bragg and Ising models, the authors introduce two essential extensions. First, the monomer concentration in solution is treated explicitly as a chemical potential term (μ = μ⁰ + RT ln c), allowing the model to describe the equilibrium between free monomers and fibrillar aggregates. Second, each peptide chain is endowed with two internal microscopic states: an α‑helical “folded” state that dominates in isolated monomers, and a β‑sheet state that participates in fibril formation. These states are represented by a binary spin variable σ_i (0 = α‑helix, 1 = β‑sheet). Adjacent chains interact via an Ising‑like coupling J σ_iσ_{i+1}, while the intrinsic energy difference between the two conformations is Δε.
The Hamiltonian reads
H = −J ∑{i}σ_iσ{i+1} + Δε ∑{i}σ_i − μ ∑{i}n_i,
where n_i indicates whether chain i is incorporated into a fibril. Because the system is one‑dimensional with nearest‑neighbour interactions, the partition function can be evaluated exactly using a 2 × 2 transfer matrix. The largest eigenvalue λ_max determines the free energy (F = −RT ln λ_max) and, through its derivatives, yields observable quantities such as the average fibril fraction, mean fibril length, heat capacity, and susceptibility.
By scanning temperature (T) and monomer concentration (c), the authors construct a two‑dimensional phase diagram. At low T and low c the α‑helical monomer dominates; increasing either T or c drives a cooperative transition to a β‑sheet‑rich fibrillar phase. The transition is marked by a discontinuity in the first derivative of the free energy with respect to T or μ, reflecting the experimentally observed “critical concentration” and “critical temperature” for amyloid formation. The steepness of the transition is controlled by the coupling J (cooperativity) and the intrinsic bias Δε; larger J produces a sharper, more switch‑like aggregation, whereas smaller J yields a gradual crossover.
The theoretical predictions are compared with published experimental data on Aβ peptide aggregation, including Thioflavin‑T fluorescence assays, electron‑microscopy measurements of fibril morphology, and sedimentation analyses that quantify monomer versus fibril populations. The model reproduces the experimentally determined aggregation temperature (≈ 310 K) and the concentration threshold (≈ 30–50 µM) at which fibril formation accelerates dramatically. Although the parameters J and Δε are tuned within physically reasonable ranges, the qualitative agreement persists across a broad parameter space, demonstrating that the essential physics of amyloid aggregation is captured without resorting to computationally intensive molecular dynamics simulations.
Key contributions of the work are: (1) a rigorously solvable framework that simultaneously accounts for microscopic conformational states and macroscopic solution conditions; (2) an explicit inclusion of monomer concentration, enabling a thermodynamic description of the critical concentration phenomenon; (3) a clear mapping between model parameters and experimentally observable cooperativity, providing a bridge between theory and measurement; and (4) a platform that can be readily extended to incorporate additional complexities such as solvent effects, heterogeneous peptide sequences, or nucleation barriers.
In conclusion, the authors deliver a concise yet powerful model that elucidates the balance between α‑helical monomers and β‑sheet fibrils in β‑amyloid systems. The analytical tractability allows rapid exploration of phase behavior and parameter sensitivity, offering a valuable tool for guiding experimental design, interpreting aggregation assays, and ultimately informing the development of therapeutic agents that modulate amyloid fibril formation. Future work may embed the present framework into higher‑dimensional lattice models or couple it with kinetic schemes to address nucleation kinetics and fibril elongation dynamics, thereby advancing our quantitative understanding of amyloid diseases.
Comments & Academic Discussion
Loading comments...
Leave a Comment