Toolbox model of evolution of metabolic pathways on networks of arbitrary topology
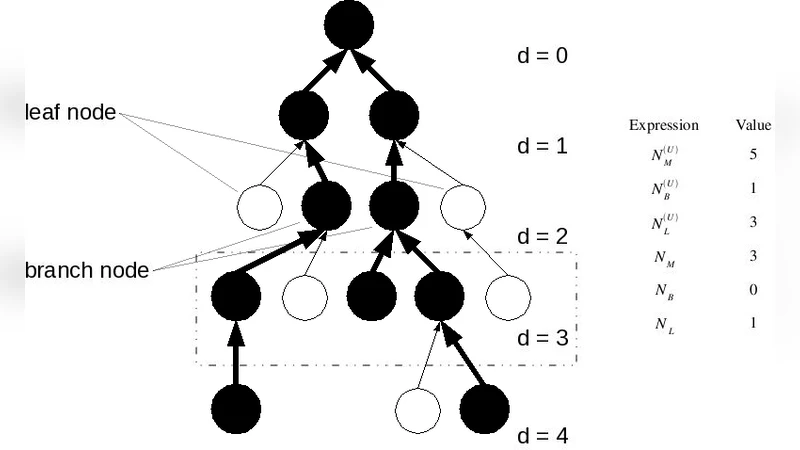
In prokaryotic genomes the number of transcriptional regulators is known to quadratically scale with the total number of protein-coding genes. Toolbox model was recently proposed to explain this scaling for metabolic enzymes and their regulators. According to its rules the metabolic network of an organism evolves by horizontal transfer of pathways from other species. These pathways are part of a larger “universal” network formed by the union of all species-specific networks. It remained to be understood, however, how the topological properties of this universal network influence the scaling law of functional content of genomes. In this study we answer this question by first analyzing the scaling properties of the toolbox model on arbitrary tree-like universal networks. We mathematically prove that the critical branching topology, in which the average number of upstream neighbors of a node is equal to one, is both necessary and sufficient for the quadratic scaling. Conversely, the toolbox model on trees with exponentially expanding, supercritical topology is characterized by the linear scaling with logarithmic corrections. We further generalize our model to include reactions with multiple substrates/products as well as branched or cyclic metabolic pathways. Unlike the original model the new version employs evolutionary optimized pathways with the smallest number of reactions necessary to achieve their metabolic tasks. Numerical simulations of this most realistic model on the universal network from the KEGG database again produced approximately quadratic scaling. Our results demonstrate why, in spite of their “small-world” topology, real-life metabolic networks are characterized by a broad distribution of pathway lengths and sizes of metabolic regulons in regulatory networks.
💡 Research Summary
The paper tackles a long‑standing observation in prokaryotic genomics: the number of transcriptional regulators grows roughly quadratically with the total number of protein‑coding genes. The “toolbox model” was previously proposed to explain this relationship for metabolic enzymes and their regulators, positing that an organism’s metabolic network expands by acquiring whole pathways through horizontal gene transfer from other species. All species‑specific pathways together form a “universal” metabolic network, and the model’s scaling behavior depends on the topology of this universal network.
The authors first formalize the model on arbitrary tree‑like universal networks. They define a node as a metabolite and consider the average number of upstream neighbors ⟨k⟩ (the branching factor). Using rigorous combinatorial arguments, they prove that when ⟨k⟩ = 1 – the critical branching case – the number of regulators R scales quadratically with the number of enzymes E (R ∝ E²). In this regime each newly acquired pathway introduces almost entirely new reactions, so a distinct regulator is required for each new enzyme set, yielding the observed quadratic law. Conversely, for super‑critical trees (⟨k⟩ > 1) the network expands exponentially; many enzymes become part of already existing pathways, and the scaling becomes linear with a logarithmic correction (R ∝ E log E). Thus the topology of the universal network is both necessary and sufficient for the quadratic scaling.
Recognizing that real metabolic networks are not simple trees, the authors extend the model to include reactions with multiple substrates and products, as well as branched and cyclic pathways. They introduce an “evolutionary optimization” principle: for any metabolic task the organism selects the pathway with the smallest possible number of reactions that still accomplishes the task. This constraint mimics natural selection for metabolic efficiency and limits pathway length while preserving functional diversity.
To test the extended model, the authors constructed a universal network from the KEGG database, comprising thousands of metabolites and reactions and exhibiting small‑world characteristics. They then simulated the acquisition of pathways according to the optimized toolbox rules. Despite the presence of cycles, multi‑substrate reactions, and a broad distribution of pathway lengths, the simulations reproduced an approximately quadratic relationship between enzyme count and regulator count, matching empirical genomic data. Moreover, the distribution of pathway lengths and regulon sizes in the simulated genomes mirrored the wide spread observed in real organisms, explaining why small‑world metabolic graphs still generate diverse regulatory demands.
The study’s contributions are threefold. First, it provides a rigorous mathematical link between universal network topology and the scaling law of metabolic regulons. Second, it demonstrates that even when realistic biochemical complexities are added, the quadratic scaling persists, validating the toolbox model beyond its original simplistic assumptions. Third, it offers an evolutionary rationale: natural selection may drive universal metabolic networks toward a critical branching architecture because such a structure balances the acquisition of novel functions with manageable regulatory overhead.
Implications extend to synthetic biology and genome engineering. When designing artificial microbes, targeting a critical branching topology for the metabolic backbone could ensure that the number of required regulators grows predictably with pathway expansion, simplifying circuit design. Future work could incorporate stochastic horizontal transfer rates, selective pressures on pathway utility, and dynamic regulation to further refine the model. Overall, the paper convincingly bridges network theory, evolutionary biology, and systems genomics to explain a fundamental scaling law in prokaryotic metabolism.
Comments & Academic Discussion
Loading comments...
Leave a Comment