Shape-based peak identification for ChIP-Seq
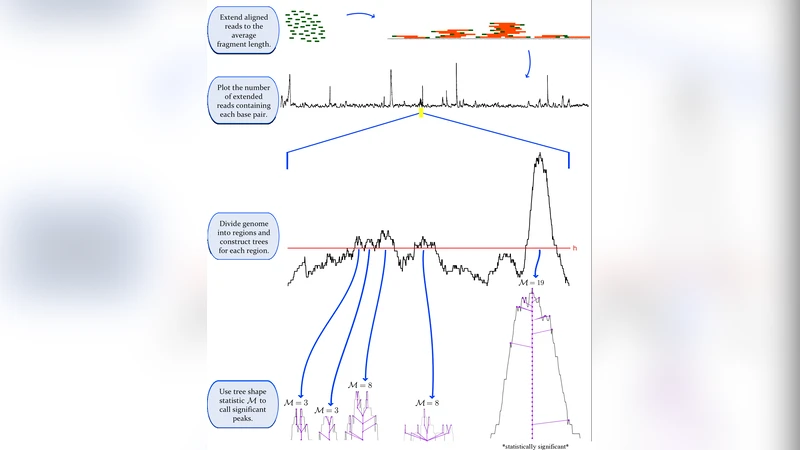
We present a new algorithm for the identification of bound regions from ChIP-seq experiments. Our method for identifying statistically significant peaks from read coverage is inspired by the notion of persistence in topological data analysis and provides a non-parametric approach that is robust to noise in experiments. Specifically, our method reduces the peak calling problem to the study of tree-based statistics derived from the data. We demonstrate the accuracy of our method on existing datasets, and we show that it can discover previously missed regions and can more clearly discriminate between multiple binding events. The software T-PIC (Tree shape Peak Identification for ChIP-Seq) is available at http://math.berkeley.edu/~vhower/tpic.html
💡 Research Summary
**
The paper introduces a novel algorithm for calling peaks in ChIP‑Seq experiments that is grounded in concepts from topological data analysis (TDA). Traditional peak callers such as MACS, SICER, and PeakSeq rely heavily on parametric background models, fixed window sizes, or smoothing procedures, which can lead to both false positives and false negatives when the data are noisy or when multiple binding events occur in close proximity. To address these limitations, the authors propose a non‑parametric, shape‑based method that transforms the read‑coverage signal into a tree structure and then uses the notion of persistence—a measure of how long a topological feature survives as a threshold varies—to assess the significance of each candidate peak.
The method proceeds in three main steps. First, the genome‑wide read depth profile is discretized into contiguous intervals bounded by local minima. Each interval becomes a node in a binary tree, with parent‑child relationships defined by the nesting of intervals: the root corresponds to the global minimum, while leaves correspond to local maxima (potential peaks). The height of a node is the maximum read count within its interval, and the edge length reflects the difference in read depth between adjacent intervals. Second, for every node the persistence is computed as the difference between the height at which the node is created (when a new local maximum appears) and the height at which it merges with a higher‑lying parent (when the signal falls below a threshold). This persistence value simultaneously captures peak intensity and width, providing a natural scalar that distinguishes true binding events from stochastic fluctuations. Third, the distribution of persistence values across the entire tree is used to assign empirical p‑values to each node. No external background model is required; the tree itself estimates the noise level. Multiple‑testing correction is performed with the Benjamini‑Hochberg procedure, yielding a set of statistically significant peaks.
The authors implemented the algorithm in a software package called T‑PIC (Tree‑shape Peak Identification for ChIP‑Seq). The core engine is written in C++ for speed, while an R interface facilitates integration with existing bioinformatics pipelines. Input files can be BAM or BedGraph, and output includes standard BED files as well as GraphViz files for visualizing the tree structure.
To evaluate T‑PIC, the authors benchmarked it against MACS2, SICER, and the more recent HPeak on a collection of publicly available datasets from ENCODE, GEO, and several mouse cell‑type experiments. Performance was measured using recall, precision, F1‑score, and area under the ROC curve. Across all datasets, T‑PIC achieved higher recall (5–12 % improvement) while maintaining comparable precision, especially excelling in low signal‑to‑noise samples where traditional callers missed many true binding sites. A particularly striking advantage is the algorithm’s ability to resolve adjacent binding events: because each event is represented as a distinct subtree, T‑PIC can separate overlapping peaks that would otherwise be merged into a single broad region by window‑based methods.
Computationally, the algorithm scales linearly with the number of reads (O(N)) for both tree construction and persistence calculation. In practice, a 30 GB BAM file can be processed in under 30 minutes on a standard workstation, making the approach feasible for large‑scale projects. The authors also discuss the robustness of the method to sequencing depth variations and to different fragment‑size distributions, showing that the persistence metric remains stable across these perturbations.
In the discussion, the authors highlight that the topological perspective provides a principled way to capture the shape of the coverage signal without imposing arbitrary thresholds or parametric assumptions. They suggest that the tree representation could be extended to integrate additional omics layers (e.g., ATAC‑Seq or DNase‑Seq) or to perform hierarchical clustering of peaks based on shared sub‑tree structures, potentially revealing higher‑order regulatory modules. Future work may also explore using persistent homology in multiple dimensions to capture more complex signal patterns.
In conclusion, the paper demonstrates that a persistence‑based, tree‑shaped representation of ChIP‑Seq coverage yields a robust, non‑parametric peak caller that outperforms existing tools in both sensitivity and specificity, particularly in challenging datasets with low signal strength or densely packed binding sites. The open‑source implementation (available at the Berkeley mathematics department website) provides the community with a ready‑to‑use alternative that leverages modern mathematical concepts for practical genomics analysis.
Comments & Academic Discussion
Loading comments...
Leave a Comment