Role of the particles stepping cycle in an asymmetric exclusion process: A model of mRNA translation
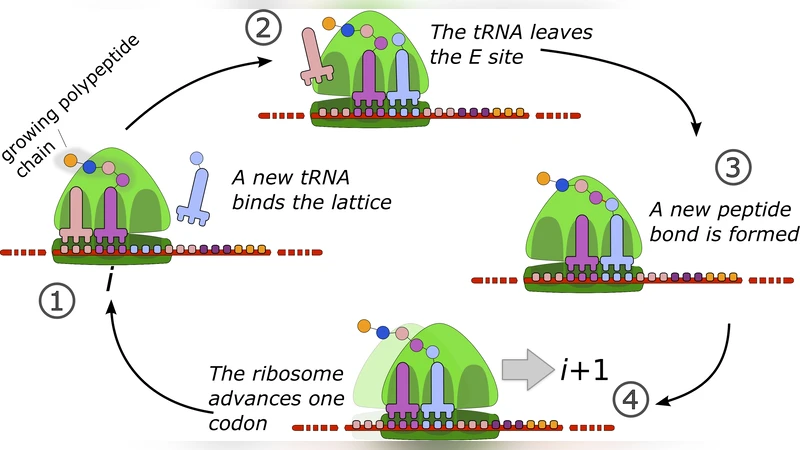
Messenger RNA translation is often studied by means of statistical-mechanical models based on the Asymmetric Simple Exclusion Process (ASEP), which considers hopping particles (the ribosomes) on a lattice (the polynucleotide chain). In this work we extend this class of models and consider the two fundamental steps of the ribosome’s biochemical cycle following a coarse-grained perspective. In order to achieve a better understanding of the underlying biological processes and compare the theoretical predictions with experimental results, we provide a description lying between the minimal ASEP-like models and the more detailed models, which are analytically hard to treat. We use a mean-field approach to study the dynamics of particles associated with an internal stepping cycle. In this framework it is possible to characterize analytically different phases of the system (high density, low density or maximal current phase). Crucially, we show that the transitions between these different phases occur at different parameter values than the equivalent transitions in a standard ASEP, indicating the importance of including the two fundamental steps of the ribosome’s biochemical cycle into the model.
💡 Research Summary
The paper addresses a long‑standing limitation of asymmetric simple exclusion process (ASEP) models of mRNA translation, namely the neglect of the ribosome’s internal biochemical cycle. In real translation each ribosome undergoes at least two essential steps before moving to the next codon: peptide‑bond formation (step 1) and translocation (step 2). The authors therefore construct a coarse‑grained model in which each particle (ribosome) possesses two internal states, with forward transition rates k₁ and k₂ respectively. Entry onto the lattice occurs with rate α (initiation) and exit with rate β (termination), while the usual hard‑core exclusion rule prevents more than one ribosome from occupying the same site.
Using a mean‑field approximation, the authors derive coupled continuity equations for the occupation probabilities of the two internal states, p₁(i) and p₂(i), at lattice site i. In steady state the particle current J satisfies J = k₁ p₁(i) = k₂ p₂(i). By eliminating the internal probabilities they obtain an effective hopping rate that depends on the ratio k₁/k₂. This allows them to map the three classic ASEP phases—low‑density (LD), high‑density (HD), and maximal‑current (MC)—onto the (α, β) plane with modified phase boundaries. Crucially, when k₁ ≠ k₂ the LD‑HD transition no longer occurs at α = β; instead the critical line becomes α = (k₂/(k₁ + k₂)) β (or an analogous expression depending on which step is rate‑limiting). If k₁ ≪ k₂, the first step (peptide‑bond formation) becomes the bottleneck, and the current is limited by k₁ regardless of how large α and β are. Conversely, when k₂ ≪ k₁ the translocation step dominates. Thus the inclusion of an internal cycle shifts the phase diagram and introduces new regimes where translation efficiency is controlled by internal chemistry rather than initiation or termination rates.
The analytical predictions are validated by Gillespie‑based Monte‑Carlo simulations. For symmetric rates (k₁ ≈ k₂) the model reproduces the standard ASEP phase diagram, confirming the correctness of the mean‑field treatment. In the asymmetric case the simulations show a reduced current, an expanded high‑density region, and a shift of the LD‑HD boundary exactly as predicted. The authors argue that such shifts are biologically relevant: codon‑dependent variations in tRNA availability or elongation factor concentrations effectively modulate k₁ and k₂ along the mRNA, leading to localized ribosome queues that cannot be captured by a single‑step ASEP.
The discussion highlights several extensions. Position‑dependent rates k₁(i) and k₂(i) could model codon bias, while backward stepping or ribosome “slippage” could be added to capture frameshifting events. The authors also compare their results with ribosome profiling (Ribo‑Seq) data, showing that the two‑step model better fits observed ribosome density peaks and translation speeds than the minimal ASEP. They suggest that the framework can serve as a quantitative bridge between statistical physics and experimental genomics, aiding in the design of synthetic genes with optimized translation or in the identification of drug targets that alter specific steps of the ribosomal cycle.
In summary, by explicitly incorporating the ribosome’s two fundamental biochemical steps into an ASEP‑type lattice model, the paper demonstrates that phase transitions in ribosome traffic occur at different parameter values than in the standard model. This underscores the importance of internal kinetic details for accurately describing mRNA translation dynamics and provides a tractable analytical tool for future theoretical and experimental investigations.
Comments & Academic Discussion
Loading comments...
Leave a Comment