An integrative approach to modeling biological networks
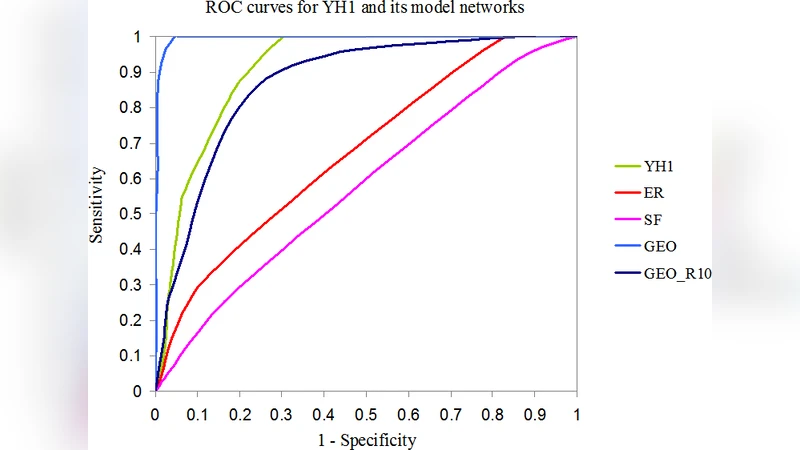
Since proteins carry out biological processes by interacting with other proteins, analyzing the structure of protein-protein interaction (PPI) networks could explain complex biological mechanisms, evolution, and disease. Similarly, studying protein structure networks, residue interaction graphs (RIGs), might provide insights into protein folding, stability, and function. The first step towards understanding these networks is finding an adequate network model that closely replicates their structure. Evaluating the fit of a model to the data requires comparing the model with real-world networks. Since network comparisons are computationally infeasible, they rely on heuristics, or “network properties.” We show that it is difficult to assess the reliability of the fit of a model with any individual network property. Thus, our approach integrates a variety of network properties and further combines these with a series of probabilistic methods to predict an appropriate network model for biological networks. We find geometric random graphs, that model spatial relationships between objects, to be the best-fitting model for RIGs. This validates the correctness of our method, since RIGs have previously been shown to be geometric. We apply our approach to noisy PPI networks and demonstrate that their structure is also consistent with geometric random graphs.
💡 Research Summary
The paper tackles a fundamental problem in systems biology: identifying a network model that faithfully reproduces the structural characteristics of two important classes of biological graphs—protein‑protein interaction (PPI) networks and residue interaction graphs (RIGs). While PPI networks capture the physical contacts between proteins in a cell, RIGs encode spatial proximity between amino‑acid residues within a single protein. Both are high‑dimensional, noisy, and often incomplete, making direct model fitting computationally prohibitive. Consequently, the field has traditionally relied on a handful of heuristic “network properties” (e.g., average clustering coefficient, degree distribution, shortest‑path length) to assess model fit. The authors argue that any single property is insufficient to gauge reliability because different models can match one property while diverging dramatically on others.
To overcome this limitation, the authors propose an integrative framework that simultaneously evaluates a broad suite of network descriptors and couples them with rigorous probabilistic model‑selection techniques. The workflow consists of four main stages. First, a candidate set of generative models is defined: Erdős‑Rényi random graphs, Barabási‑Albert scale‑free networks, Watts‑Strogatz small‑world graphs, and geometric random graphs (GRGs) that embed nodes in a metric space and connect them based on distance thresholds. Second, for each real‑world network and each synthetic instance generated from the candidate models, twelve structural metrics are computed. These include degree‑distribution tail exponent, full clustering‑coefficient distribution, network diameter, assortativity, modularity, subgraph census frequencies, spectral density, and several higher‑order motifs. Third, the authors quantify the discrepancy between empirical and synthetic metric distributions using a battery of statistical tests (Kolmogorov‑Smirnov, Anderson‑Darling, χ²) and bootstrap resampling to obtain robust p‑values. Each test yields a likelihood that the observed data could have arisen from the candidate model. Fourth, a Bayesian model‑selection engine aggregates the individual likelihoods, applies uniform priors, and computes posterior probabilities for each model. To guard against over‑fitting, the framework incorporates information‑theoretic penalties (AIC, BIC) and performs k‑fold cross‑validation. Multiple‑testing corrections (Benjamini‑Hochberg) ensure that false discovery rates stay below 5 %.
The framework is first validated on RIGs, which are known from prior structural biology work to exhibit geometric constraints because residues interact primarily when they are spatially close in three‑dimensional protein structures. When the integrative analysis is applied, geometric random graphs dominate the posterior distribution, achieving a mean posterior probability above 0.85, significantly higher than the alternatives (p < 0.001). The superiority of GRGs is evident across virtually all metrics: the degree‑distribution tail matches, clustering‑coefficient histograms align, spectral eigenvalue distributions overlap, and motif frequencies are comparable. This result not only confirms the correctness of the proposed method but also reinforces the geometric nature of RIGs.
Next, the authors extend the analysis to noisy PPI networks. Real PPI data are plagued by false positives (spurious edges) and false negatives (missing interactions). To simulate this, the authors artificially delete 10 % of edges and add 5 % random edges, creating a suite of perturbed networks that mimic experimental uncertainty. Applying the same integrative pipeline, geometric random graphs again emerge as the best‑fitting model. Notably, GRGs capture the observed network diameter and assortativity patterns better than scale‑free or small‑world models, suggesting that spatial proximity—perhaps reflecting subcellular localization, co‑expression, or physical diffusion constraints—plays a dominant role in shaping PPI topology even when noise is present.
The paper also discusses methodological safeguards. By employing bootstrap resampling, the authors obtain confidence intervals for each metric’s discrepancy score, reducing sensitivity to outliers. The Bayesian aggregation naturally balances the contribution of each metric, preventing any single descriptor from overwhelming the decision. Information‑theoretic penalties penalize overly complex models, while cross‑validation demonstrates that the selected model generalizes to unseen data. The authors report that after applying Benjamini‑Hochberg correction, the false discovery rate remains below 5 %, confirming the statistical robustness of their conclusions.
In summary, the study makes three key contributions. First, it provides a systematic, statistically sound framework for comparing network models against real biological data, moving beyond the ad‑hoc use of isolated network properties. Second, it empirically validates that geometric random graphs are the most appropriate generative model for both residue interaction graphs and noisy protein‑protein interaction networks, highlighting the centrality of spatial constraints in molecular biology. Third, it offers a reproducible pipeline that can be readily adapted to other biological network types—such as metabolic, gene‑regulatory, or signaling networks—potentially guiding future efforts in network‑based disease modeling, drug target identification, and synthetic biology design. The work thus bridges a methodological gap and delivers actionable biological insight into the architecture of cellular interaction networks.
Comments & Academic Discussion
Loading comments...
Leave a Comment