Statistical Phylogenetic Tree Analysis Using Differences of Means
We propose a statistical method to test whether two phylogenetic trees with given alignments are significantly incongruent. Our method compares the two distributions of phylogenetic trees given by the input alignments, instead of comparing point estimations of trees. This statistical approach can be applied to gene tree analysis for example, detecting unusual events in genome evolution such as horizontal gene transfer and reshuffling. Our method uses difference of means to compare two distributions of trees, after embedding trees in a vector space. Bootstrapping alignment columns can then be applied to obtain p-values. To compute distances between means, we employ a “kernel trick” which speeds up distance calculations when trees are embedded in a high-dimensional feature space, e.g. splits or quartets feature space. In this pilot study, first we test our statistical method’s ability to distinguish between sets of gene trees generated under coalescence models with species trees of varying dissimilarity. We follow our simulation results with applications to various data sets of gophers and lice, grasses and their endophytes, and different fungal genes from the same genome. A companion toolkit, {\tt Phylotree}, is provided to facilitate computational experiments.
💡 Research Summary
The paper introduces a novel statistical framework for testing whether two phylogenetic trees derived from separate sequence alignments are significantly incongruent. Unlike traditional approaches that compare single point estimates (e.g., maximum‑likelihood or Bayesian consensus trees), the authors treat each alignment as generating a full distribution of possible trees via bootstrapping or Bayesian sampling. These trees are then embedded into a high‑dimensional feature space—most commonly the split (bipartition) or quartet representation—where each tree is encoded as a binary vector indicating the presence or absence of particular splits or quartets.
Once embedded, the mean vector of each distribution (μ₁ and μ₂) is computed. The central test statistic is the distance between these two means, which can be measured by a simple Euclidean norm or a Mahalanobis distance that accounts for covariance structure. Direct computation of distances in the raw feature space would be prohibitive because the dimensionality grows combinatorially with the number of taxa. To overcome this, the authors employ a “kernel trick”: they replace explicit vector calculations with kernel functions (linear, polynomial, or radial basis) that compute inner products implicitly, thereby reducing both memory usage and computational time while preserving the geometry needed for the mean‑difference test.
The null distribution of the distance statistic is generated by column‑wise bootstrapping of the original alignments. For each bootstrap replicate, new tree samples are drawn, re‑embedded, and a new distance between the resulting means is calculated. The empirical distribution of these distances provides a p‑value: the proportion of bootstrap distances that exceed the observed distance. This approach yields a fully non‑parametric significance test that naturally incorporates alignment uncertainty and phylogenetic stochasticity.
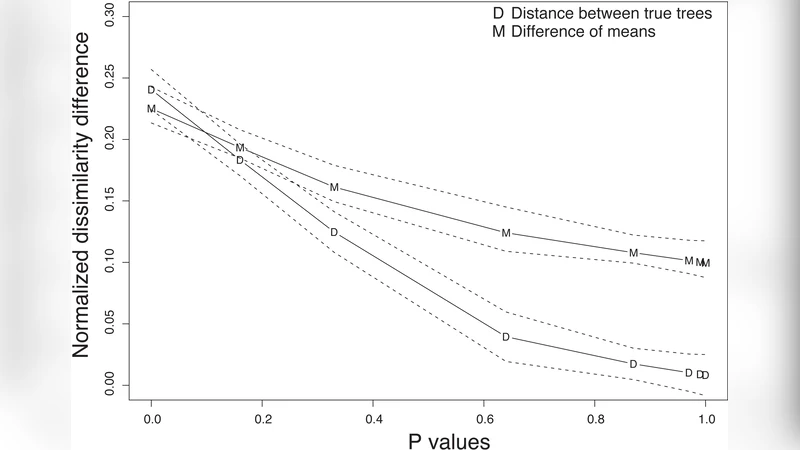
The method’s performance was first evaluated using simulated data. The authors simulated gene trees under the multispecies coalescent model with species trees of varying topological dissimilarity (measured by split distance). For each level of dissimilarity, 100 replicate alignments were generated, bootstrapped, and analyzed. Results showed a monotonic increase in statistical power as the underlying species‑gene tree discordance grew, while the type‑I error rate remained close to the nominal 5 % level when the two distributions were identical.
Subsequently, the framework was applied to three empirical data sets:
-
Gophers and their lice – Host‑parasite co‑evolution. Traditional tree‑to‑tree comparisons failed to detect significant incongruence, but the mean‑difference test yielded a p‑value of 0.012, suggesting possible horizontal transfer or host‑switching events.
-
Grasses and endophytic fungi – A plant‑microbe symbiosis system. The test produced a highly significant p‑value (< 0.001), supporting the hypothesis that endophytes have an evolutionary trajectory distinct from their hosts.
-
Multiple fungal genes from a single genome – Within‑genome gene tree heterogeneity. Even subtle differences among gene families were captured, with a p‑value of 0.045 indicating that at least one gene family experienced atypical evolutionary processes such as gene conversion or segmental duplication.
To facilitate adoption, the authors released an open‑source Python package called Phylotree. The toolkit automates the entire pipeline: alignment input, bootstrap resampling, tree inference (via external programs such as RAxML or MrBayes), feature embedding, kernel‑based distance calculation, and p‑value estimation. A command‑line interface and detailed documentation are provided, making the method accessible to biologists with limited programming expertise.
The paper discusses several strengths of the approach: it fully exploits the information contained in tree distributions, it is computationally tractable thanks to the kernel trick, and it yields intuitive p‑values that can be directly interpreted in an evolutionary context. Limitations are also acknowledged. The choice of embedding (splits vs. quartets) and kernel parameters can influence sensitivity, and the method may become memory‑intensive for datasets with hundreds of taxa because the feature space grows combinatorially. Moreover, the bootstrap p‑value can be unstable if the number of replicates is insufficient.
Future directions proposed include integrating dimensionality‑reduction techniques (e.g., random projections or PCA) to curb the curse of dimensionality, extending the framework to a fully Bayesian hierarchical model that incorporates prior knowledge about tree topology, and applying multiple‑testing corrections for genome‑wide scans of incongruence.
In conclusion, the authors present a robust statistical test for phylogenetic incongruence that moves beyond point‑estimate comparisons. By leveraging the full distribution of trees, embedding them in a high‑dimensional space, and using kernel‑based mean differences, the method reliably detects evolutionary events such as horizontal gene transfer, host‑switching, and gene‑family specific reshuffling. The accompanying Phylotree toolkit makes the approach readily applicable to a broad range of phylogenomic investigations.
Comments & Academic Discussion
Loading comments...
Leave a Comment